

Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome
- PMID: 28378819
- PMCID: PMC5477372
- DOI: 10.1038/ejhg.2017.45
Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome
Abstract
Glucose transporter type 1 deficiency syndrome (GLUT1DS) is a neurometabolic disorder with a complex phenotypic spectrum but simple biomarkers in cerebrospinal fluid. The disorder is caused by impaired glucose transport into the brain resulting from variants in SCL2A1. In 10% of GLUT1DS patients, a genetic diagnosis can not be made. Using whole-genome sequencing, we identified a de novo 5'-UTR variant in SLC2A1, generating a novel translation initiation codon, severely compromising SLC2A1 function. This finding expands our understanding of the disease mechanisms underlying GLUT1DS and encourages further in-depth analysis of SLC2A1 non-coding regions in patients without variants in the coding region.
Conflict of interest statement
JK has received speakers honoraria and travel costs from Nutricia GmbH, Eerlangen, Germany and Vitaflo Pharma GmbH, Bad Homburg, Germany. The remaining authors declare no conflict of interest.
Figures
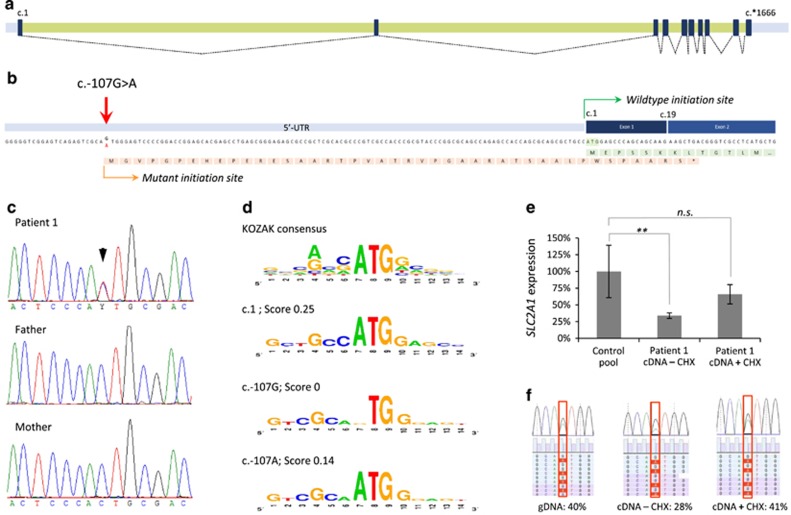
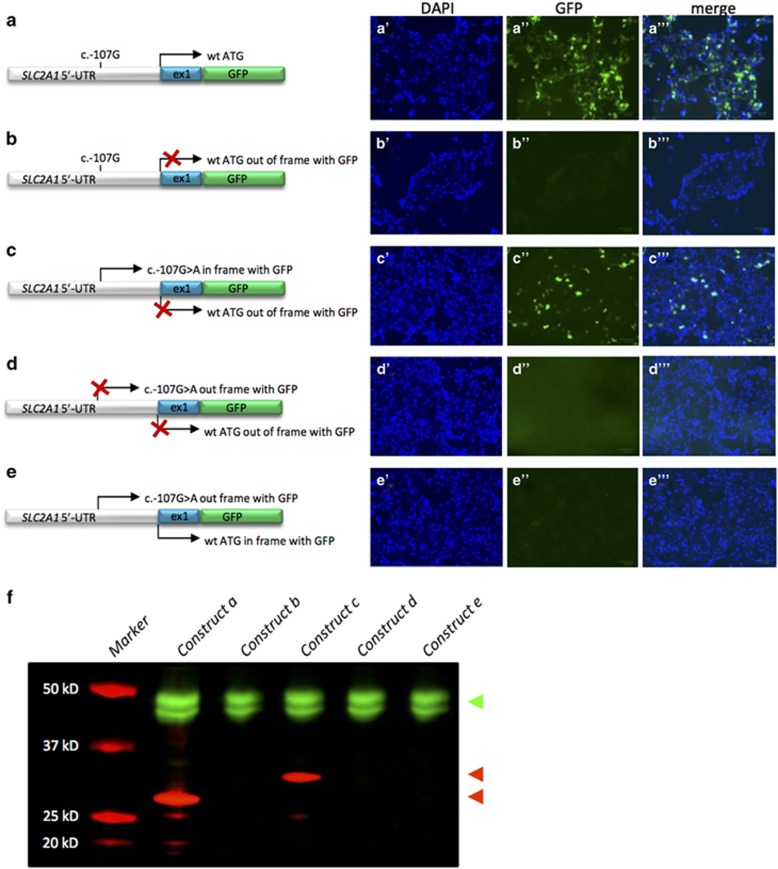
References
-
- De Vivo DC, Trifiletti RR, Jacobson RI et al: Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991; 325: 703–709. - PubMed
-
- Seidner G, Alvarez MG, Yeh JI et al: GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 1998; 18: 188–191. - PubMed
-
- Leen WG, Klepper J, Verbeek MM et al: Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010; 133: 655–670. - PubMed
-
- Hully M, Vuillaumier-Barrot S, Le Bizec C et al: From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet 2015; 58: 443–454. - PubMed
-
- Liu YC, Lee JW, Bellows ST et al: Evaluation of non-coding variation in GLUT1 deficiency. Dev Med Child Neurol 2016; 58: 1295–1302. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
