

Kart: a divide-and-conquer algorithm for NGS read alignment
- PMID: 28379292
- PMCID: PMC5860120
- DOI: 10.1093/bioinformatics/btx189
Kart: a divide-and-conquer algorithm for NGS read alignment
Abstract
Motivation: Next-generation sequencing (NGS) provides a great opportunity to investigate genome-wide variation at nucleotide resolution. Due to the huge amount of data, NGS applications require very fast and accurate alignment algorithms. Most existing algorithms for read mapping basically adopt seed-and-extend strategy, which is sequential in nature and takes much longer time on longer reads.
Results: We develop a divide-and-conquer algorithm, called Kart, which can process long reads as fast as short reads by dividing a read into small fragments that can be aligned independently. Our experiment result indicates that the average size of fragments requiring the more time-consuming gapped alignment is around 20 bp regardless of the original read length. Furthermore, it can tolerate much higher error rates. The experiments show that Kart spends much less time on longer reads than other aligners and still produce reliable alignments even when the error rate is as high as 15%.
Availability and implementation: Kart is available at https://github.com/hsinnan75/Kart/ .
Contact: hsu@iis.sinica.edu.tw.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2017. Published by Oxford University Press.
Figures

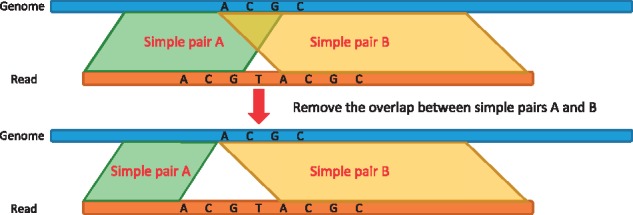
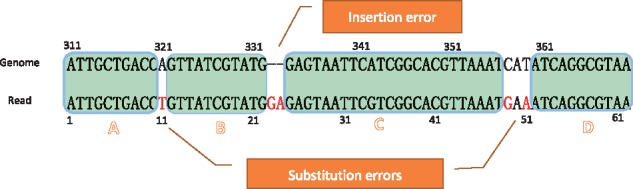
References
-
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
