

BEESEM: estimation of binding energy models using HT-SELEX data
- PMID: 28379348
- PMCID: PMC5860122
- DOI: 10.1093/bioinformatics/btx191
BEESEM: estimation of binding energy models using HT-SELEX data
Abstract
Motivation: Characterizing the binding specificities of transcription factors (TFs) is crucial to the study of gene expression regulation. Recently developed high-throughput experimental methods, including protein binding microarrays (PBM) and high-throughput SELEX (HT-SELEX), have enabled rapid measurements of the specificities for hundreds of TFs. However, few studies have developed efficient algorithms for estimating binding motifs based on HT-SELEX data. Also the simple method of constructing a position weight matrix (PWM) by comparing the frequency of the preferred sequence with single-nucleotide variants has the risk of generating motifs with higher information content than the true binding specificity.
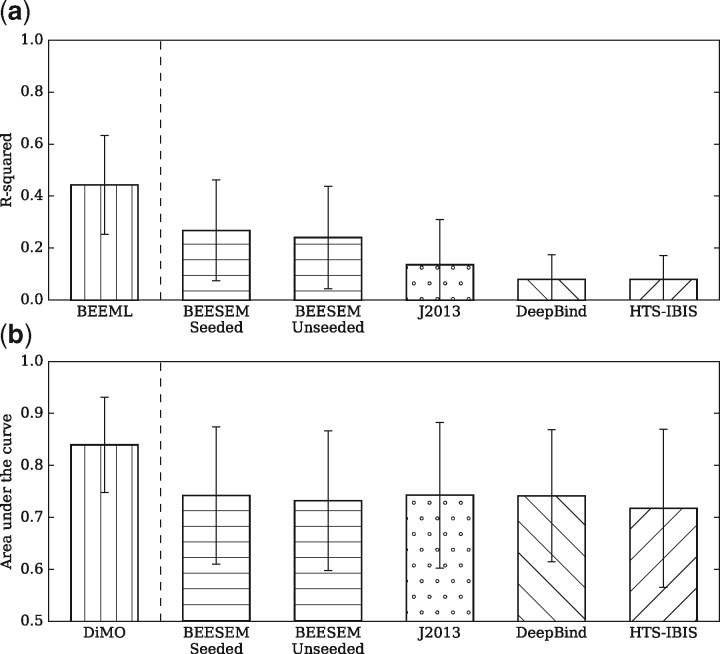
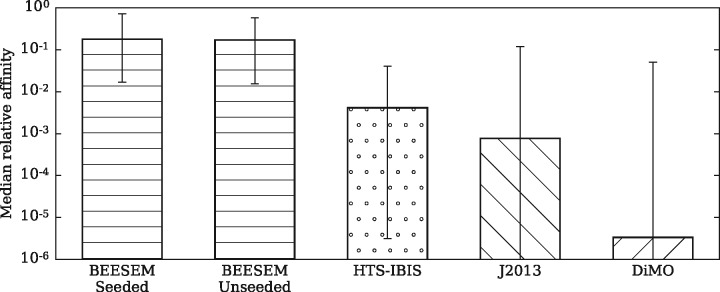
Results: We developed an algorithm called BEESEM that builds on a comprehensive biophysical model of protein-DNA interactions, which is trained using the expectation maximization method. BEESEM is capable of selecting the optimal motif length and calculating the confidence intervals of estimated parameters. By comparing BEESEM with the published motifs estimated using the same HT-SELEX data, we demonstrate that BEESEM provides significant improvements. We also evaluate several motif discovery algorithms on independent PBM and ChIP-seq data. BEESEM provides significantly better fits to in vitro data, but its performance is similar to some other methods on in vivo data under the criterion of the area under the receiver operating characteristic curve (AUROC). This highlights the limitations of the purely rank-based AUROC criterion. Using quantitative binding data to assess models, however, demonstrates that BEESEM improves on prior models.
Availability and implementation: Freely available on the web at http://stormo.wustl.edu/resources.html .
Contact: stormo@wustl.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author (2017). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com
Figures
Similar articles
-
A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data.Nucleic Acids Res. 2014 Apr;42(8):e63. doi: 10.1093/nar/gku117. Epub 2014 Feb 5. Nucleic Acids Res. 2014. PMID: 24500199 Free PMC article.
-
High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions.PLoS Comput Biol. 2010 Sep 9;6(9):e1000916. doi: 10.1371/journal.pcbi.1000916. PLoS Comput Biol. 2010. PMID: 20838582 Free PMC article.
-
Optimally choosing PWM motif databases and sequence scanning approaches based on ChIP-seq data.BMC Bioinformatics. 2015 May 1;16:140. doi: 10.1186/s12859-015-0573-5. BMC Bioinformatics. 2015. PMID: 25927199 Free PMC article.
-
A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data.Biol Direct. 2014 Feb 20;9:4. doi: 10.1186/1745-6150-9-4. Biol Direct. 2014. PMID: 24555784 Free PMC article. Review.
-
Protein binding microarrays for the characterization of DNA-protein interactions.Adv Biochem Eng Biotechnol. 2007;104:65-85. doi: 10.1007/10_025. Adv Biochem Eng Biotechnol. 2007. PMID: 17290819 Free PMC article. Review.
Cited by
-
DNA binding specificity of all four Saccharomyces cerevisiae forkhead transcription factors.Nucleic Acids Res. 2023 Jun 23;51(11):5621-5633. doi: 10.1093/nar/gkad372. Nucleic Acids Res. 2023. PMID: 37177995 Free PMC article.
-
PRIESSTESS: interpretable, high-performing models of the sequence and structure preferences of RNA-binding proteins.Nucleic Acids Res. 2022 Oct 28;50(19):e111. doi: 10.1093/nar/gkac694. Nucleic Acids Res. 2022. PMID: 36018788 Free PMC article.
-
Prediction of cooperative homeodomain DNA binding sites from high-throughput-SELEX data.Nucleic Acids Res. 2023 Jul 7;51(12):6055-6072. doi: 10.1093/nar/gkad318. Nucleic Acids Res. 2023. PMID: 37114997 Free PMC article.
-
SelexGLM differentiates androgen and glucocorticoid receptor DNA-binding preference over an extended binding site.Genome Res. 2018 Jan;28(1):111-121. doi: 10.1101/gr.222844.117. Epub 2017 Dec 1. Genome Res. 2018. PMID: 29196557 Free PMC article.
-
Sharing DNA-binding information across structurally similar proteins enables accurate specificity determination.Nucleic Acids Res. 2020 Jan 24;48(2):e9. doi: 10.1093/nar/gkz1087. Nucleic Acids Res. 2020. PMID: 31777934 Free PMC article.
References
-
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. - PubMed
-
- Atherton J. et al. (2012) A model for sequential evolution of ligands by exponential enrichment (SELEX) data. Ann. Appl. Stat., 6, 928–949.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
