

Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions
- PMID: 28380383
- PMCID: PMC5578459
- DOI: 10.1016/j.cmet.2017.03.016
Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions
Abstract
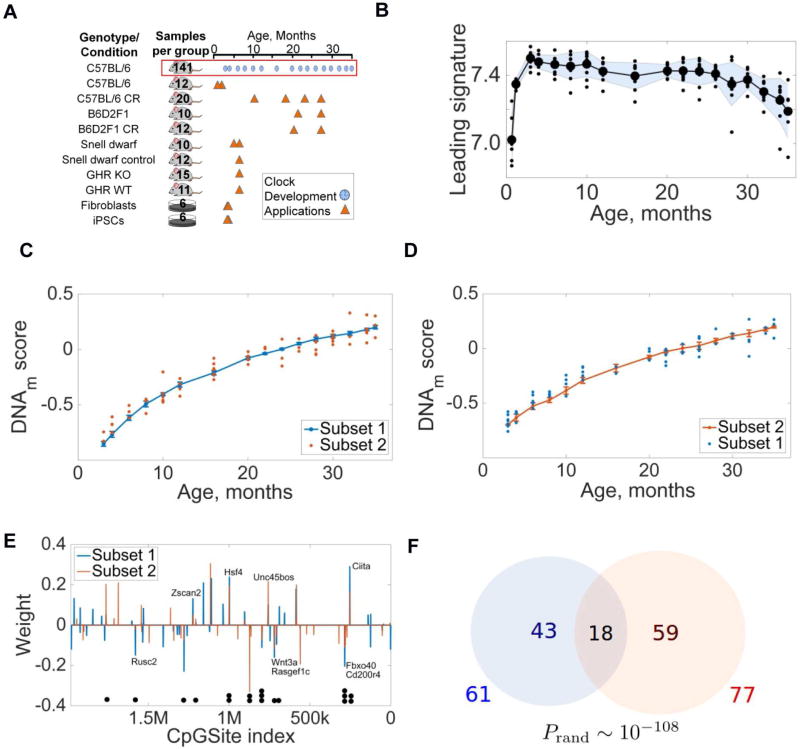
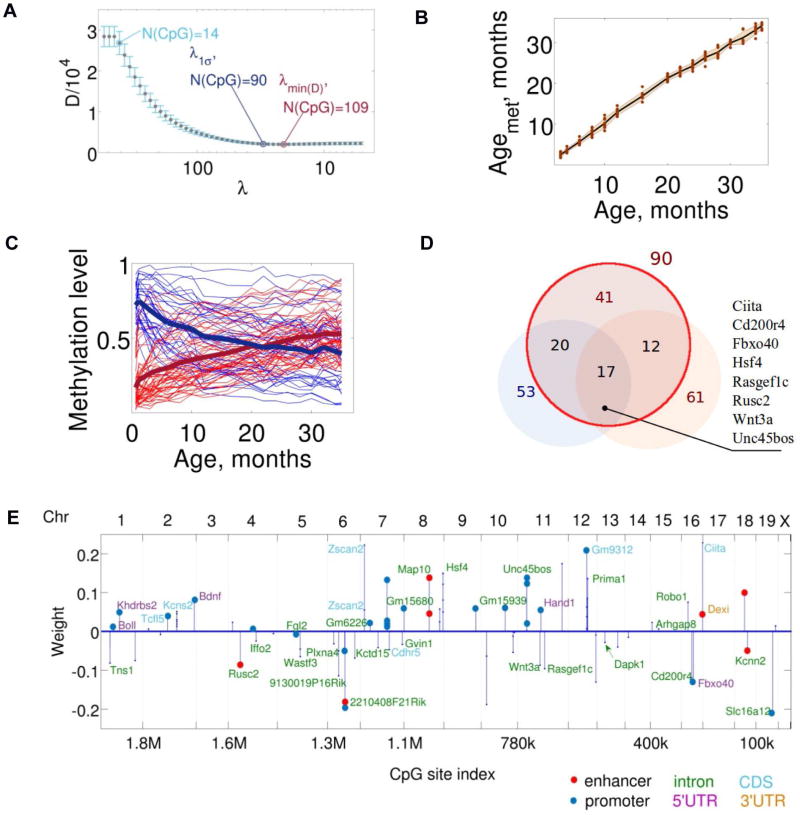
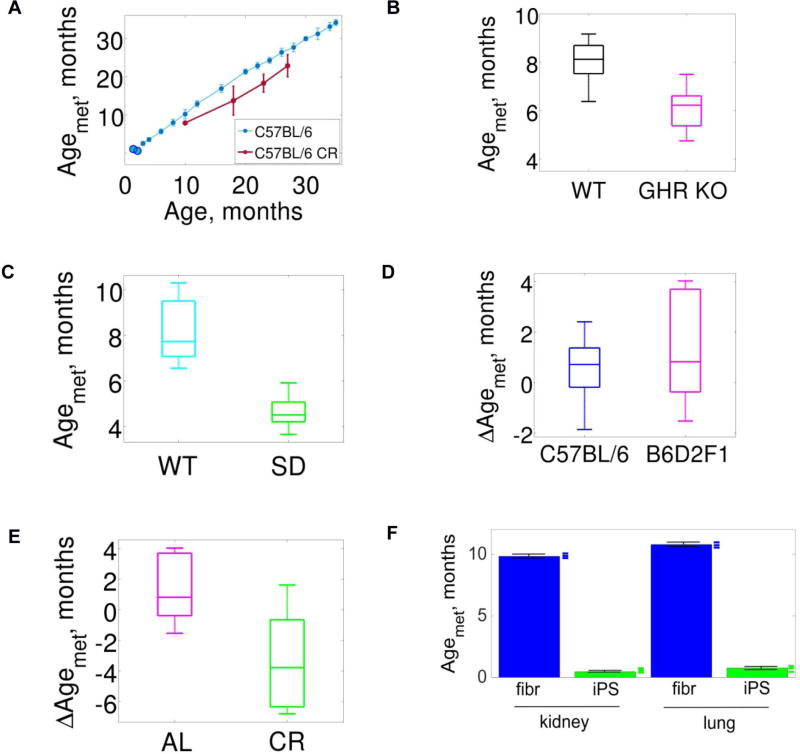
The DNA methylation levels of certain CpG sites are thought to reflect the pace of human aging. Here, we developed a robust predictor of mouse biological age based on 90 CpG sites derived from partial blood DNA methylation profiles. The resulting clock correctly determines the age of mouse cohorts, detects the longevity effects of calorie restriction and gene knockouts, and reports rejuvenation of fibroblast-derived iPSCs. The data show that mammalian DNA methylomes are characterized by CpG sites that may represent the organism's biological age. They are scattered across the genome, they are distinct in human and mouse, and their methylation gradually changes with age. The clock derived from these sites represents a biomarker of aging and can be used to determine the biological age of organisms and evaluate interventions that alter the rate of aging.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Antoulas A, Sorensen D. Approximation of large-scale dynamical systems: an overview. Int. J. Appl. Math. Comput. Sci. 2001;5:1093–1121.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
