

Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure
- PMID: 28381504
- PMCID: PMC9675292
- DOI: 10.1530/JOE-16-0679
Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure
Abstract
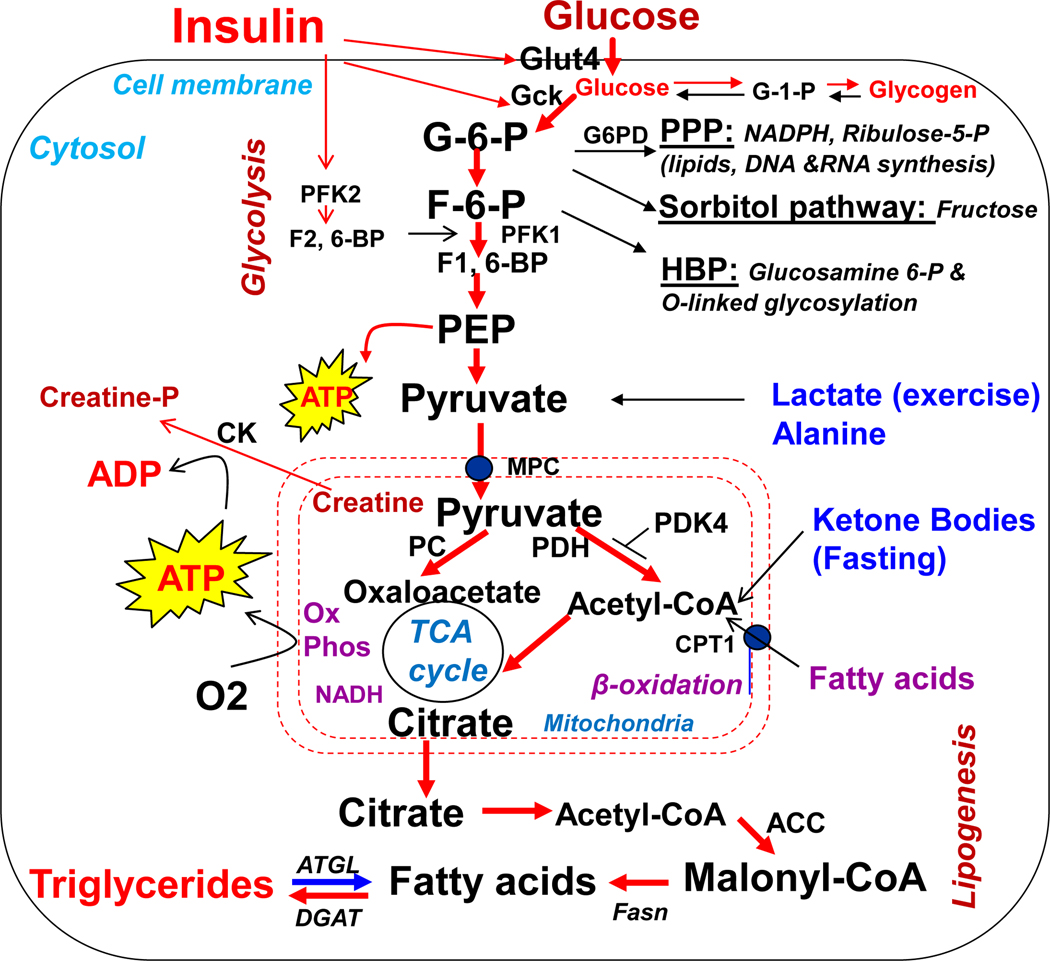
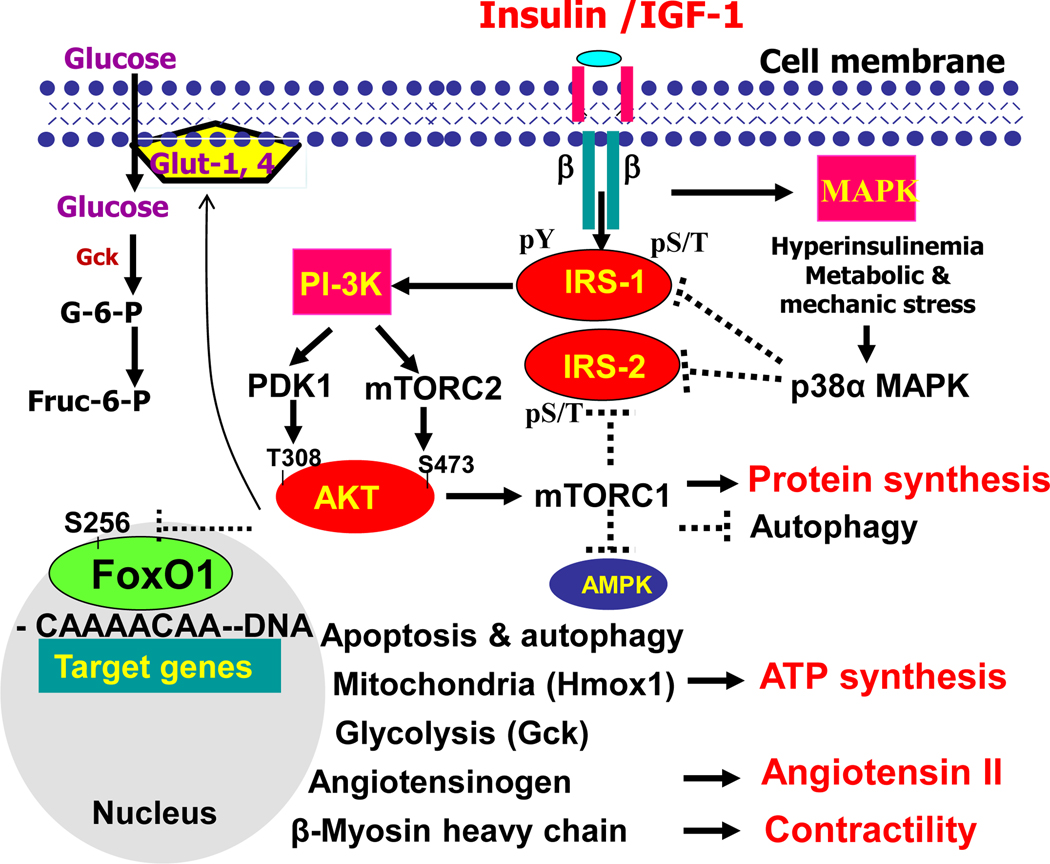
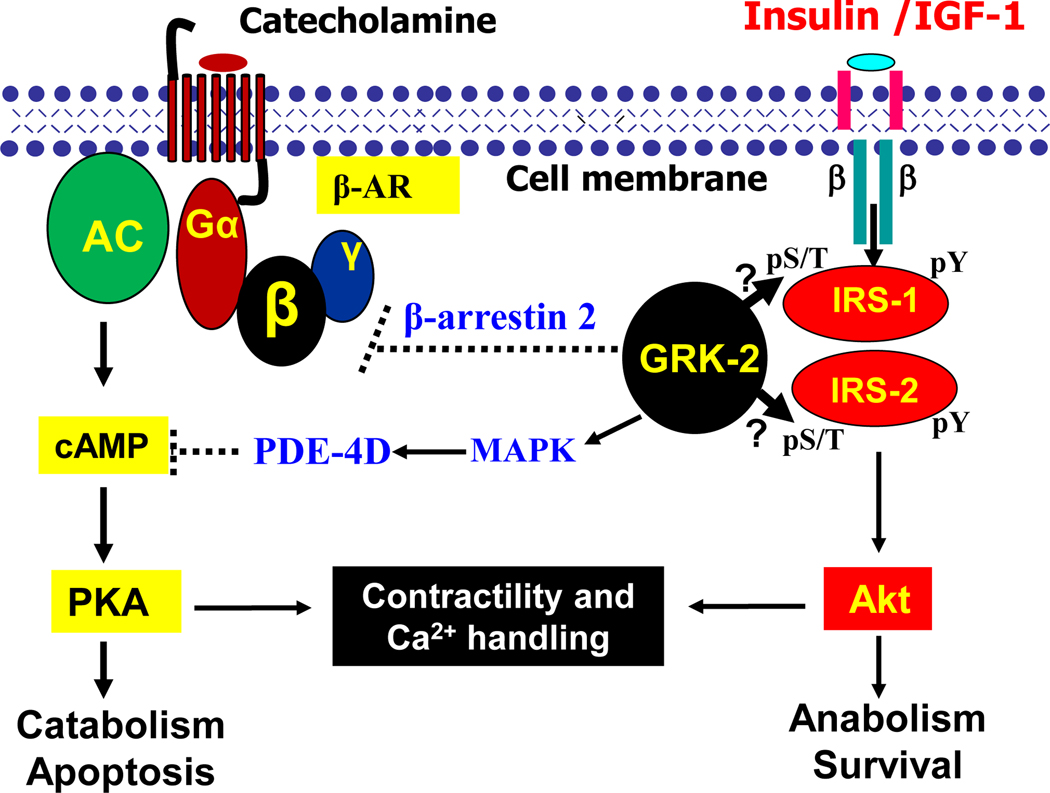
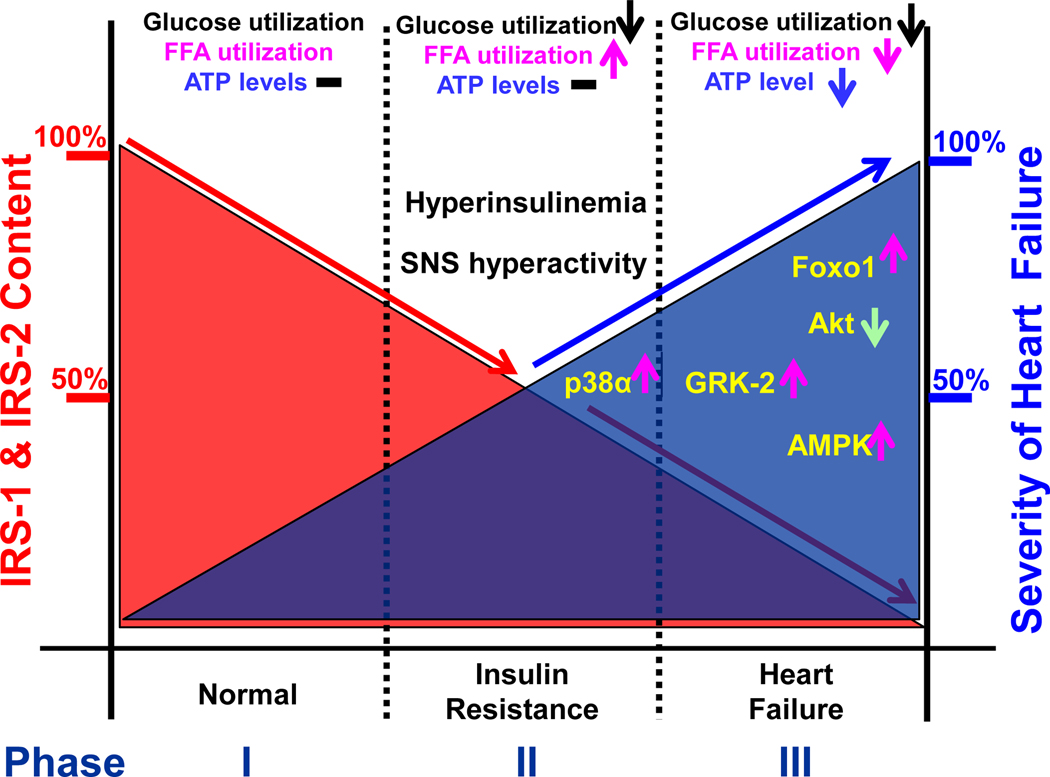
The heart is an insulin-dependent and energy-consuming organ in which insulin and nutritional signaling integrates to the regulation of cardiac metabolism, growth and survival. Heart failure is highly associated with insulin resistance, and heart failure patients suffer from the cardiac energy deficiency and structural and functional dysfunction. Chronic pathological conditions, such as obesity and type 2 diabetes mellitus, involve various mechanisms in promoting heart failure by remodeling metabolic pathways, modulating cardiac energetics and impairing cardiac contractility. Recent studies demonstrated that insulin receptor substrates 1 and 2 (IRS-1,-2) are major mediators of both insulin and insulin-like growth factor-1 (IGF-1) signaling responsible for myocardial energetics, structure, function and organismal survival. Importantly, the insulin receptor substrates (IRS) play an important role in the activation of the phosphatidylinositide-3-dependent kinase (PI-3K) that controls Akt and Foxo1 signaling cascade, regulating the mitochondrial function, cardiac energy metabolism and the renin-angiotensin system. Dysregulation of this branch in signaling cascades by insulin resistance in the heart through the endocrine system promotes heart failure, providing a novel mechanism for diabetic cardiomyopathy. Therefore, targeting this branch of IRS→PI-3K→Foxo1 signaling cascade and associated pathways may provide a fundamental strategy for the therapeutic and nutritional development in control of metabolic and cardiovascular diseases. In this review, we focus on insulin signaling and resistance in the heart and the role energetics play in cardiac metabolism, structure and function.
Keywords: forkhead transcription factor Foxo1; heart failure; insulin receptor substrates; insulin resistance.
© 2017 Society for Endocrinology.
Conflict of interest statement
Declaration of Interest
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review reported.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
