

Discovery of Novel Small-Molecule Inhibitors of LIM Domain Kinase for Inhibiting HIV-1
- PMID: 28381571
- PMCID: PMC5469273
- DOI: 10.1128/JVI.02418-16
Discovery of Novel Small-Molecule Inhibitors of LIM Domain Kinase for Inhibiting HIV-1
Abstract
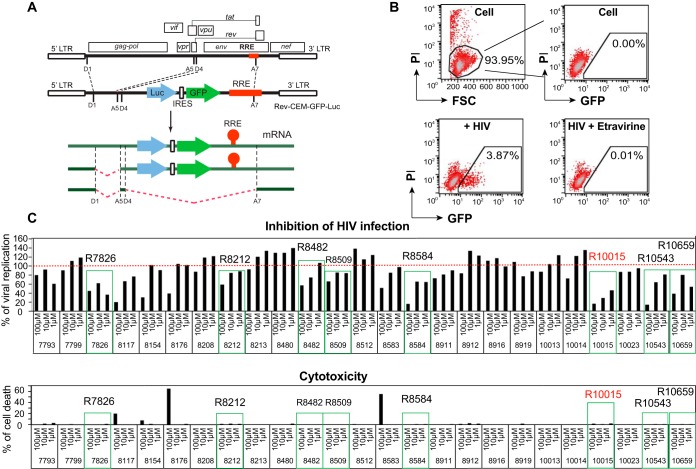
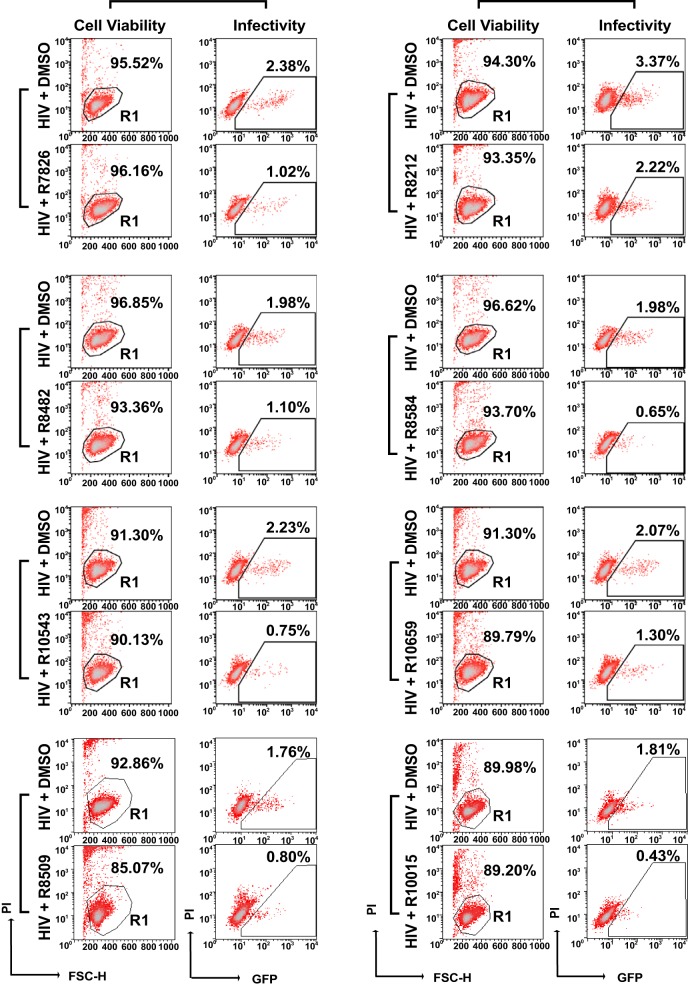
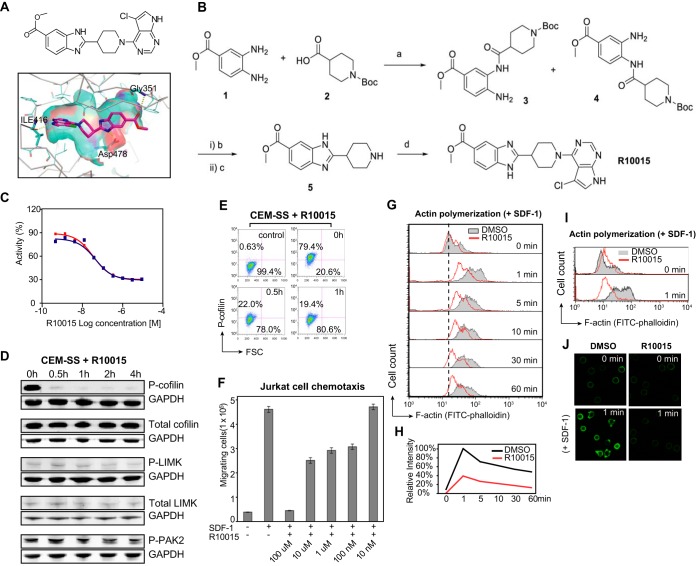
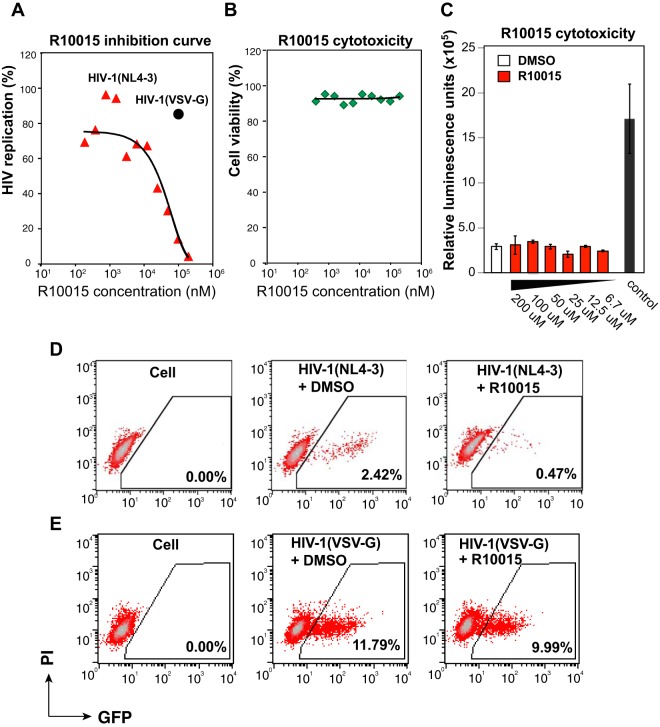
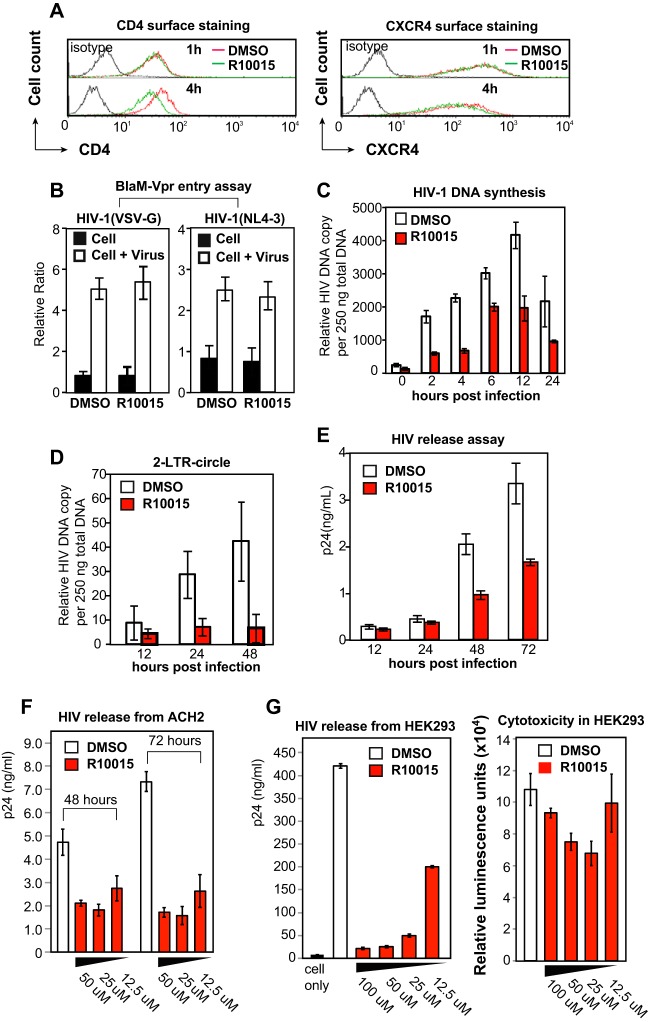
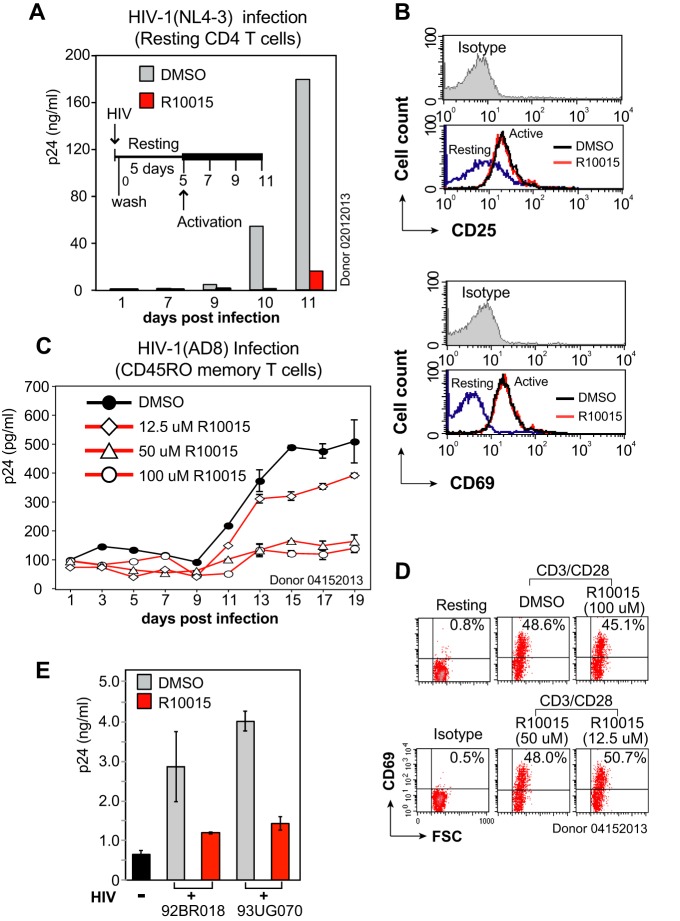
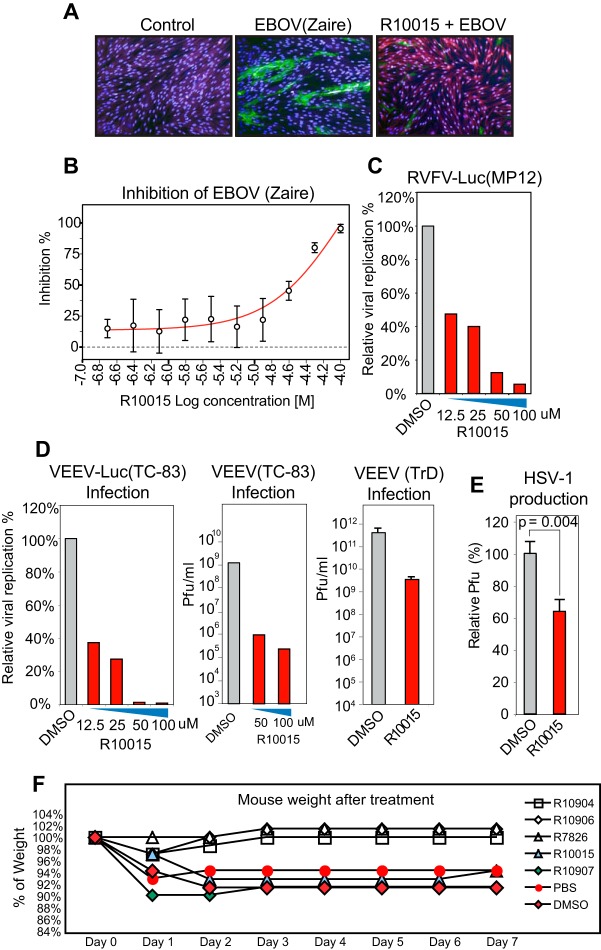
A dynamic actin cytoskeleton is necessary for viral entry, intracellular migration, and virion release. For HIV-1 infection, during entry, the virus triggers early actin activity by hijacking chemokine coreceptor signaling, which activates a host dependency factor, cofilin, and its kinase, the LIM domain kinase (LIMK). Although knockdown of human LIM domain kinase 1 (LIMK1) with short hairpin RNA (shRNA) inhibits HIV infection, no specific small-molecule inhibitor of LIMK has been available. Here, we describe the design and discovery of novel classes of small-molecule inhibitors of LIMK for inhibiting HIV infection. We identified R10015 as a lead compound that blocks LIMK activity by binding to the ATP-binding pocket. R10015 specifically blocks viral DNA synthesis, nuclear migration, and virion release. In addition, R10015 inhibits multiple viruses, including Zaire ebolavirus (EBOV), Rift Valley fever virus (RVFV), Venezuelan equine encephalitis virus (VEEV), and herpes simplex virus 1 (HSV-1), suggesting that LIMK inhibitors could be developed as a new class of broad-spectrum antiviral drugs.IMPORTANCE The actin cytoskeleton is a structure that gives the cell shape and the ability to migrate. Viruses frequently rely on actin dynamics for entry and intracellular migration. In cells, actin dynamics are regulated by kinases, such as the LIM domain kinase (LIMK), which regulates actin activity through phosphorylation of cofilin, an actin-depolymerizing factor. Recent studies have found that LIMK/cofilin are targeted by viruses such as HIV-1 for propelling viral intracellular migration. Although inhibiting LIMK1 expression blocks HIV-1 infection, no highly specific LIMK inhibitor is available. This study describes the design, medicinal synthesis, and discovery of small-molecule LIMK inhibitors for blocking HIV-1 and several other viruses and emphasizes the feasibility of developing LIMK inhibitors as broad-spectrum antiviral drugs.
Keywords: CXCR4; Ebola virus; LIM domain kinase; Rift Valley fever virus; Venezuelan equine encephalitis virus; actin; cofilin; cytoskeleton; herpes simplex virus; human immunodeficiency virus.
Copyright © 2017 Yi et al.
Figures
References
-
- Nowak M. 1990. HIV mutation rate. Nature 347:522. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
