

Direct determination of diploid genome sequences
- PMID: 28381613
- PMCID: PMC5411770
- DOI: 10.1101/gr.214874.116
Direct determination of diploid genome sequences
Erratum in
-
Corrigendum: Direct determination of diploid genome sequences.Genome Res. 2018 Apr;28(4):606.1. doi: 10.1101/gr.235812.118. Genome Res. 2018. PMID: 29610250 Free PMC article. No abstract available.
Abstract
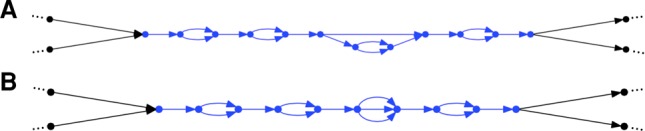
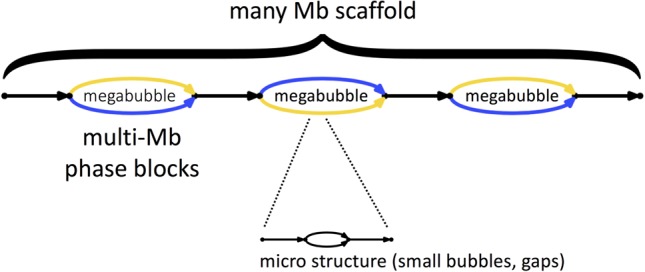
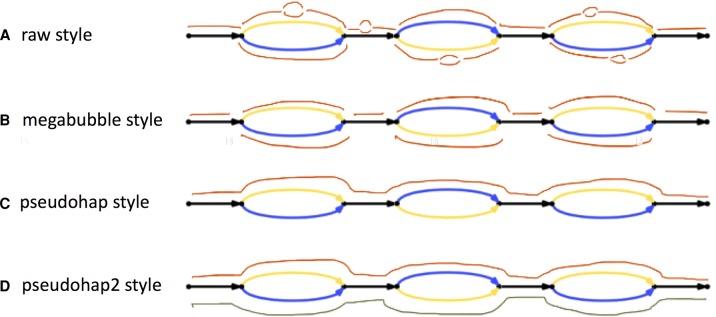
Determining the genome sequence of an organism is challenging, yet fundamental to understanding its biology. Over the past decade, thousands of human genomes have been sequenced, contributing deeply to biomedical research. In the vast majority of cases, these have been analyzed by aligning sequence reads to a single reference genome, biasing the resulting analyses, and in general, failing to capture sequences novel to a given genome. Some de novo assemblies have been constructed free of reference bias, but nearly all were constructed by merging homologous loci into single "consensus" sequences, generally absent from nature. These assemblies do not correctly represent the diploid biology of an individual. In exactly two cases, true diploid de novo assemblies have been made, at great expense. One was generated using Sanger sequencing, and one using thousands of clone pools. Here, we demonstrate a straightforward and low-cost method for creating true diploid de novo assemblies. We make a single library from ∼1 ng of high molecular weight DNA, using the 10x Genomics microfluidic platform to partition the genome. We applied this technique to seven human samples, generating low-cost HiSeq X data, then assembled these using a new "pushbutton" algorithm, Supernova. Each computation took 2 d on a single server. Each yielded contigs longer than 100 kb, phase blocks longer than 2.5 Mb, and scaffolds longer than 15 Mb. Our method provides a scalable capability for determining the actual diploid genome sequence in a sample, opening the door to new approaches in genomic biology and medicine.
© 2017 Weisenfeld et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Anantharaman T, Mishra B. 2001. False positives in genome map assembly and sequence validation. In Algorithms in bioinformatics (ed. Gascuel O, Moret BM), pp. 27–40. Springer, Berlin, Germany.
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. 2002. Recent segmental duplications in the human genome. Science 297: 1003–1007. - PubMed
-
- Bovee D, Zhou Y, Haugen E, Wu Z, Hayden HS, Gillett W, Tuzun E, Cooper GM, Sampas N, Phelps K, et al. 2008. Closing gaps in the human genome with fosmid resources generated from multiple individuals. Nat Genet 40: 96–101. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous