

The Complex Genetic Basis of Congenital Heart Defects
- PMID: 28381817
- PMCID: PMC5715472
- DOI: 10.1253/circj.CJ-16-1343
The Complex Genetic Basis of Congenital Heart Defects
Abstract
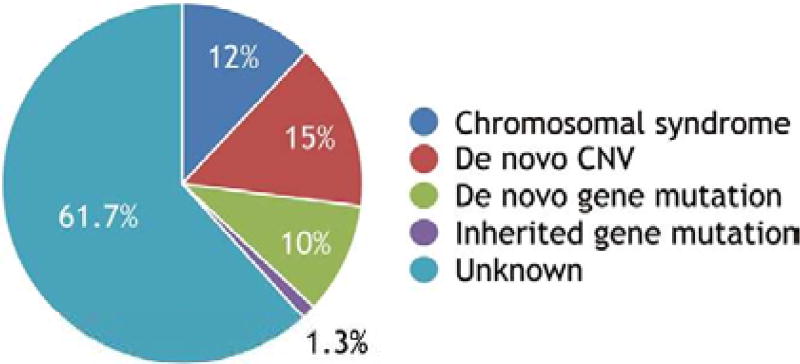
Twenty years ago, chromosomal abnormalities were the only identifiable genetic causes of a small fraction of congenital heart defects (CHD). Today, a de novo or inherited genetic abnormality can be identified as pathogenic in one-third of cases. We refer to them here as monogenic causes, insofar as the genetic abnormality has a readily detectable, large effect. What explains the other two-thirds? This review considers a complex genetic basis. That is, a combination of genetic mutations or variants that individually may have little or no detectable effect contribute to the pathogenesis of a heart defect. Genes in the embryo that act directly in cardiac developmental pathways have received the most attention, but genes in the mother that establish the gestational milieu via pathways related to metabolism and aging also have an effect. A growing body of evidence highlights the pathogenic significance of genetic interactions in the embryo and maternal effects that have a genetic basis. The investigation of CHD as guided by a complex genetic model could help estimate risk more precisely and logically lead to a means of prevention.
Keywords: Congenital heart defects; Genetics; Maternal age; Maternal effects; Modifier genes.
Figures
References
-
- Newman JR. The world of mathematics. London: George Allen and Unwin; 1956. [November 1, 2016]. Commentary on Gregor Mendel; pp. 932–936. https://archive.org/details/TheWorldOfMathematicsVolume2.
-
- Peacock TB. On malformations of the human heart. London: John Churchill and Sons; 1866. [November 1, 2016]. pp. 101–166. https://archive.org/details/onmalformationso00peac.
-
- Nora JJ. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases: The genetic-environmental interaction. Circulation. 1968;38:604–617. - PubMed
-
- Ferencz C, Neill CA, Boughman JA, Rubin JD, Brenner JI, Perry LW. Congenital cardiovascular malformations associated with chromosome abnormalities: An epidemiologic study. J Pedialr. 1989;114:79–86. - PubMed
-
- Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, et al. The contribution of chromosomal abnormalities to congenital heart defects: A population-based study. Pediatr Cardiol. 2011;32:1147–1157. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical