

Review
doi: 10.1182/blood-2017-02-765891.
Epub 2017 Apr 6.
Treating sickle cell disease by targeting HbS polymerization
Affiliations
- PMID: 28385699
- PMCID: PMC5437829
- DOI: 10.1182/blood-2017-02-765891
Item in Clipboard
Review
Treating sickle cell disease by targeting HbS polymerization
Blood.
.
Abstract
Although the root cause of sickle cell disease is the polymerization of hemoglobin S (HbS) to form fibers that make red cells less flexible, most drugs currently being assessed in clinical trials are targeting the downstream sequelae of this primary event. Less attention has been devoted to investigation of the multiple ways in which fiber formation can be inhibited. In this article, we describe the molecular rationale for 5 distinct approaches to inhibiting polymerization and also discuss progress with the few antipolymerization drugs currently in clinical trials.
Figures
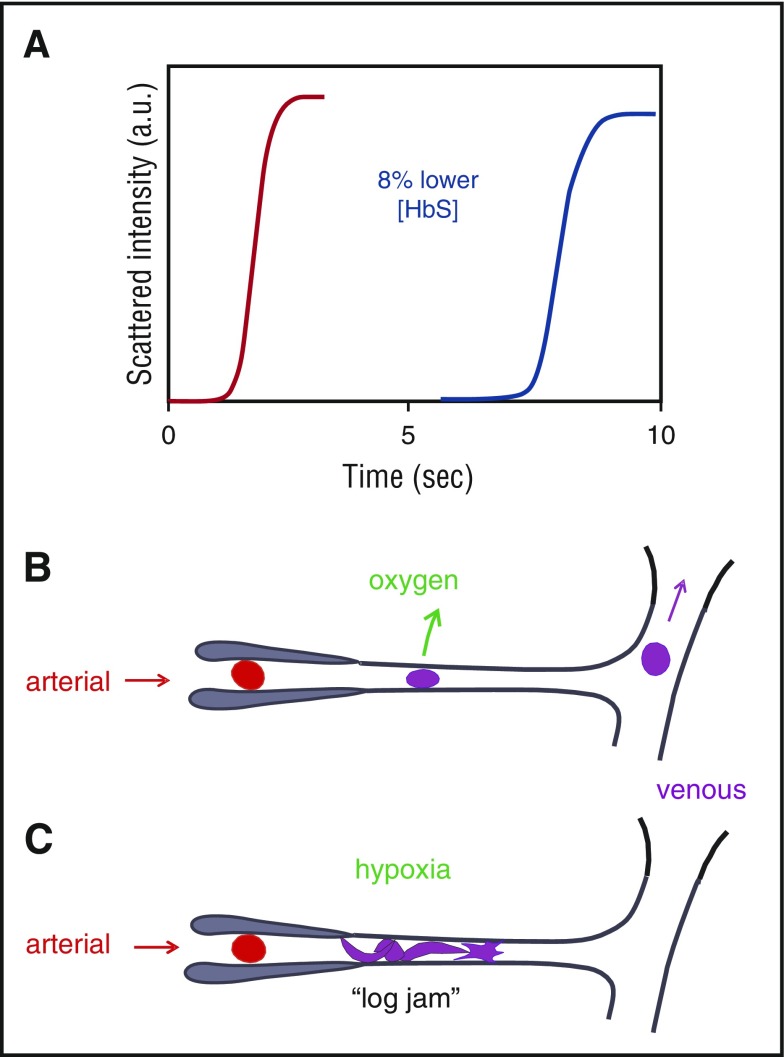
Connection between kinetics and pathophysiology. (A) Schematic of kinetic progress curve for polymerization occurring on the seconds time scale measured by light scattering due to fiber formation. Before the appearance of fibers, there is a delay (lag phase). The delay time is extraordinarily sensitive to HbS concentration, depending on the 30th power of the concentration., Such a large exponent means that a decrease of only 8% in the HbS concentration increases the delay time 10-fold. (B) Schematic of microcirculation: arteriole, capillary, and venule. The vast majority of cells escape the microcirculation before fibers form and cause cellular distortion (sickling)., (C) Schematic of vaso-occlusion. If the delay time is shorter than the transit time (or fibers have not completely dissolved upon oxygenation in the lungs and can grow without a delay,), fibers form within the small vessels and can cause vaso-occlusion. In this graphic, factors that slow the transit of red cells through the microcirculation, such as increased adherence to the vascular endothelium by damaged red cells or increased leukocytes associated with infection will increase the probability of vaso-occlusion. a.u., arbitrary units.
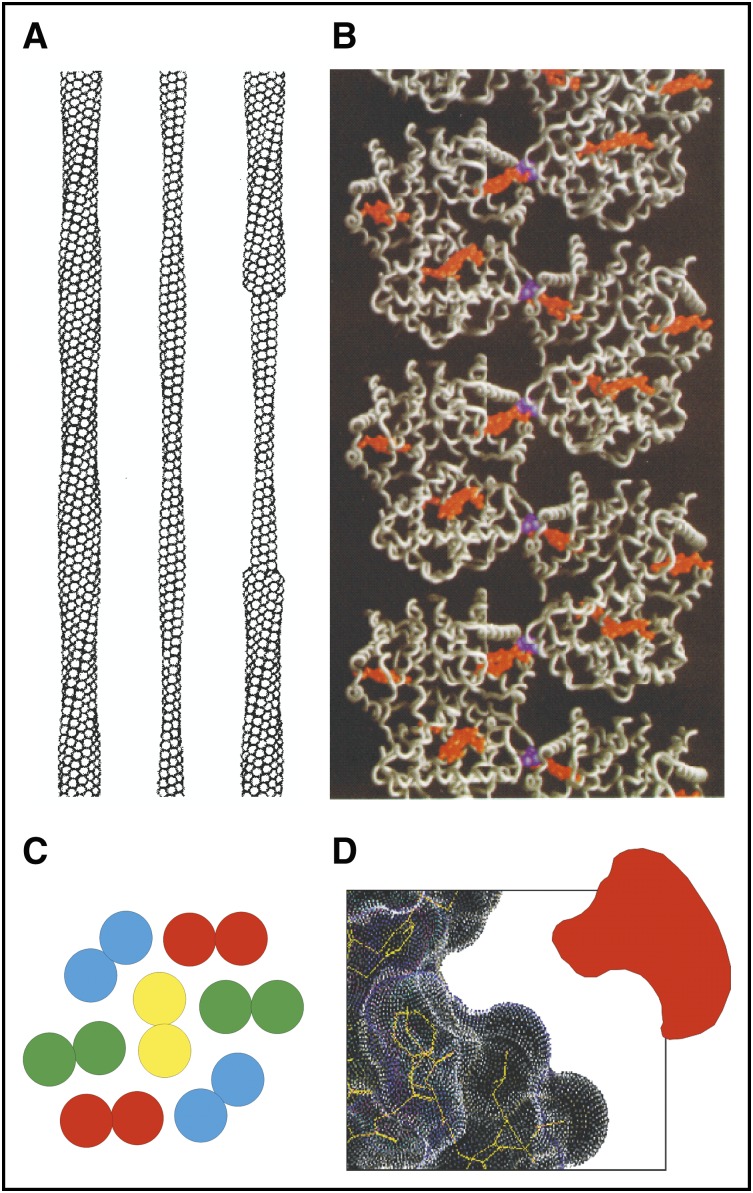
Sickle fiber structure. (A) Low-resolution structure of 14-stranded solid fiber determined by electron microscopy. Each HbS tetramer is represented as a sphere. (B) Atomic structure of deoxy-HbS determined by X-ray crystallography showing that 1 of the 2 β6 valines (purple) in each tetramer makes an intermolecular contact with an adjacent strand close to the pocket containing the hemes (orange). (C) Cross-section of sickle fiber composed of 7 double strands. (D) Cartoon of small molecule inhibitor that could fit into the shallow acceptor site for the β6 valine.
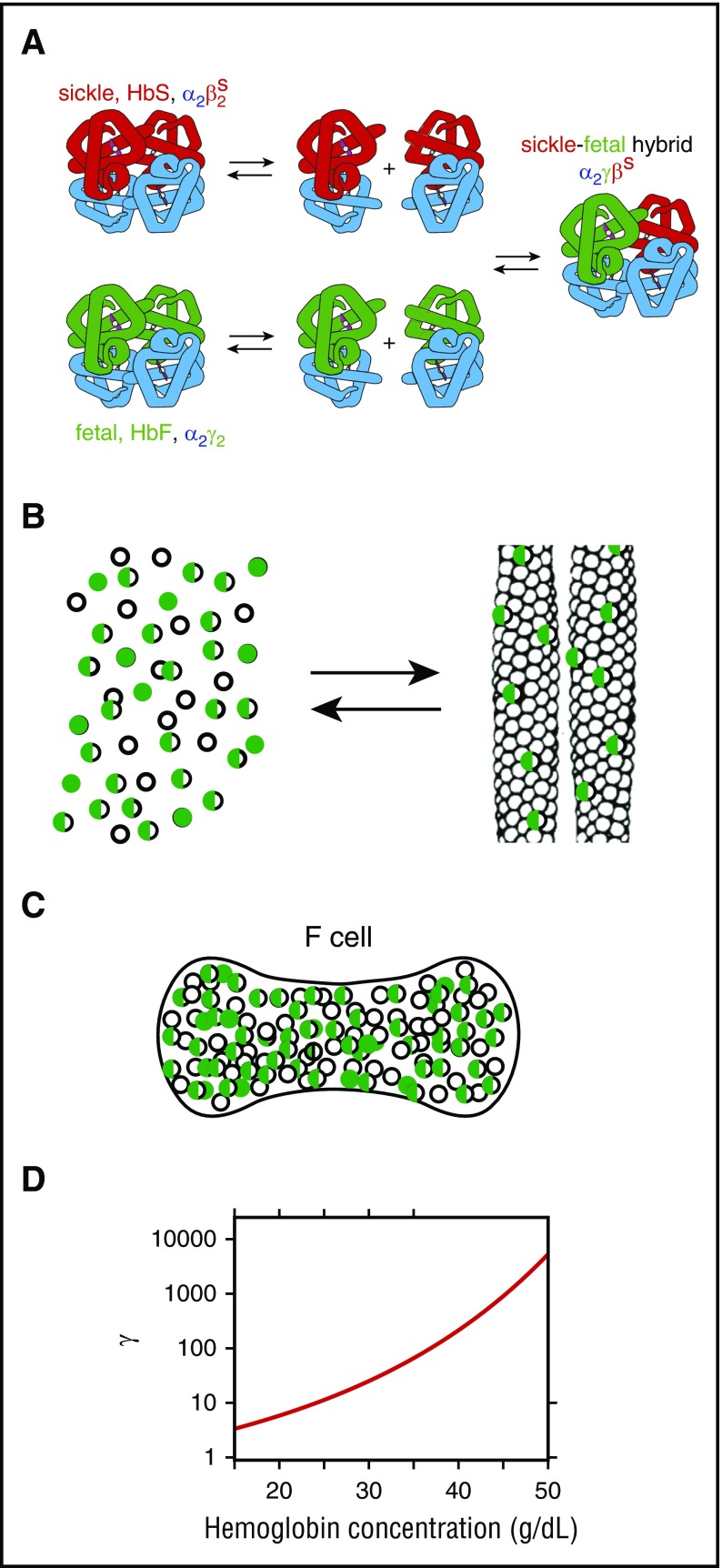
Mechanism of inhibition of polymerization by HbF. (A) Dissociation of tetramers into dimers and reassociation in mixtures of HbS and HbF result in 3 tetramers in a binomial distribution, thereby further lowering the fraction of the HbS homotetramer (α2βS2). (B) Cartoon of polymerization equilibrium in HbS-HbF mixture. As in a crystallization reaction, Hb tetramers are present in 2 phases: the solution phase (left) or the fiber phase (right). The fibers that form in these mixtures are primarily composed of the HbS homotetramers, but there is also some copolymerization of hybrid tetramers (α2βSγ) (half-green, half-empty circles)., (C) Cartoon of F cell with 30% HbF and 70% HbS. The excluded volume effect of the non-copolymerizing α2γ2 tetramer (green-filled circles) and partially copolymerizing tetramer α2βSγ (half-green, half-empty circles) increases the activity of the polymerizing α2βS2 homotetramer (empty circles). (D) Activity coefficient (γ) as a function of total Hb concentration. The dimensionless activity coefficient is the factor that multiplies the measured concentration (ie, moles per liter or grams per deciliter) to obtain the activity, which is the thermodynamically effective concentration.
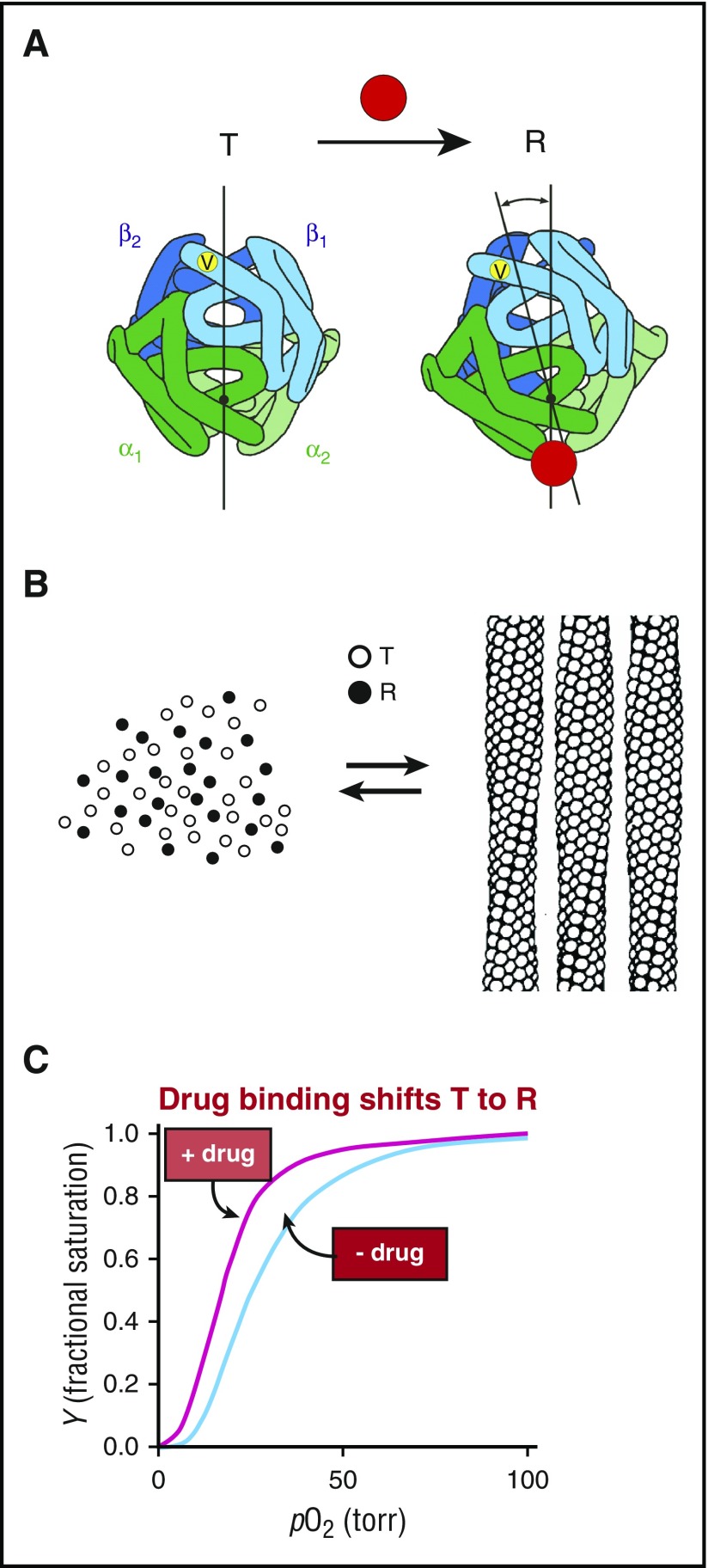
Mechanism of polymerization inhibition by increasing oxygen affinity. (A) Hemoglobin exists in a rapidly reversible equilibrium between low- and high-affinity quaternary conformations, called T and R, respectively., They differ primarily by an ∼15° relative rotation of αβ dimers. Location of β6 valine is shown as a yellow dot on the surface of the molecule. Preferential binding of a small molecule such as a drug (red circle) to R shifts the quaternary equilibrium toward R. (B) Cartoon of polymerization equilibrium. Only the T quaternary structure (empty circles) enters the fiber. R quaternary conformations (filled circles) are completely excluded. (C) Oxygen binding curves. Preferential binding of a drug to the R quaternary structure causes a left shift (increased oxygen affinity).
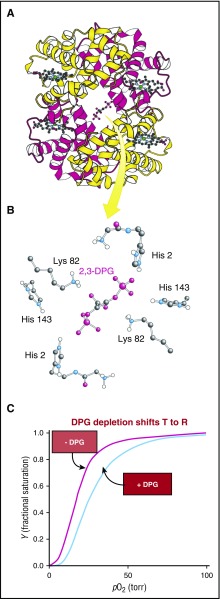
Binding of 2,3-DPG to Hb. (A-B) 2,3-DPG binds in the cleft between the β (yellow) subunits of the T quaternary structure. (C) Reduction of 2,3-DPG concentration shifts the quaternary equilibrium toward R to produce a left shift in the binding curve and increases the solubility (the concentration of Hb in the solution phase [Figures 3B and 4B, left]). Both factors decrease sickling.
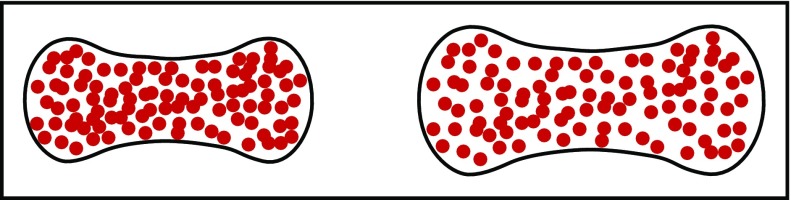
Schematic of normal and swollen red cells. A small increase in red cell volume to decrease the intracellular HbS concentration dramatically increases the delay time of sickling because of its high dependence on the HbS concentration. Even a 10% increase in cell volume is predicted to have a therapeutic effect.,
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
