

Genome-wide TOP2A DNA cleavage is biased toward translocated and highly transcribed loci
- PMID: 28385713
- PMCID: PMC5495075
- DOI: 10.1101/gr.211615.116
Genome-wide TOP2A DNA cleavage is biased toward translocated and highly transcribed loci
Abstract
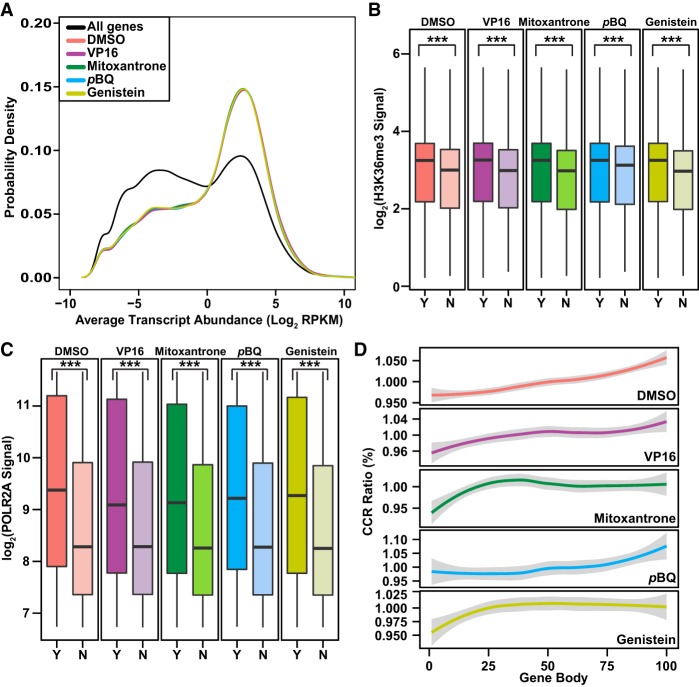
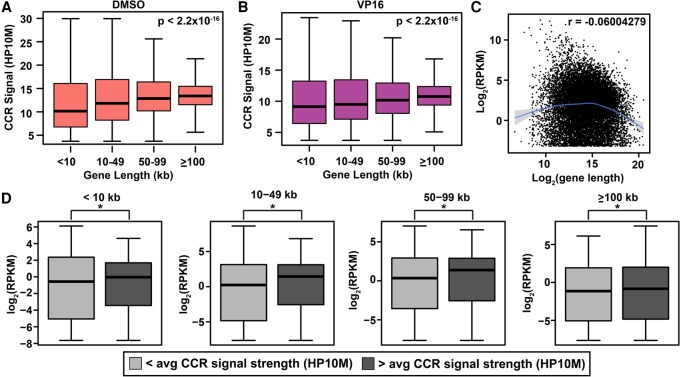
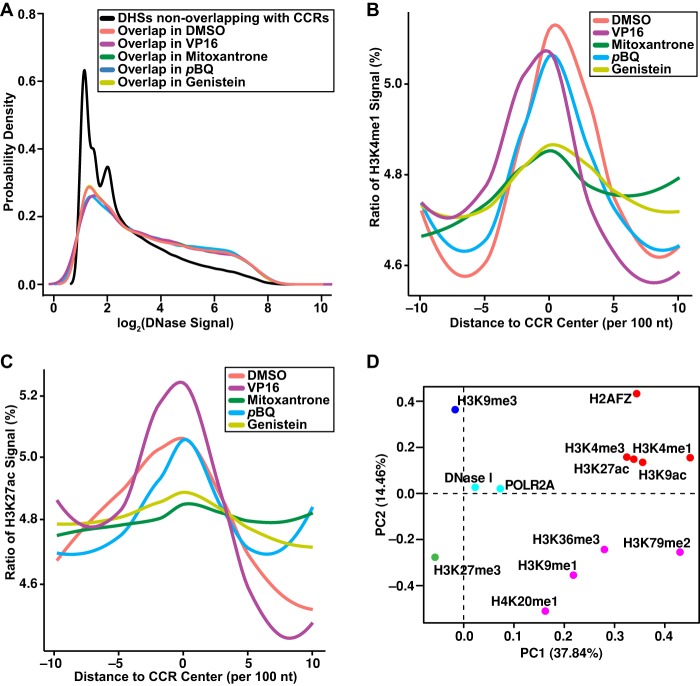
Type II topoisomerases orchestrate proper DNA topology, and they are the targets of anti-cancer drugs that cause treatment-related leukemias with balanced translocations. Here, we develop a high-throughput sequencing technology to define TOP2 cleavage sites at single-base precision, and use the technology to characterize TOP2A cleavage genome-wide in the human K562 leukemia cell line. We find that TOP2A cleavage has functionally conserved local sequence preferences, occurs in cleavage cluster regions (CCRs), and is enriched in introns and lincRNA loci. TOP2A CCRs are biased toward the distal regions of gene bodies, and TOP2 poisons cause a proximal shift in their distribution. We find high TOP2A cleavage levels in genes involved in translocations in TOP2 poison-related leukemia. In addition, we find that a large proportion of genes involved in oncogenic translocations overall contain TOP2A CCRs. The TOP2A cleavage of coding and lincRNA genes is independently associated with both length and transcript abundance. Comparisons to ENCODE data reveal distinct TOP2A CCR clusters that overlap with marks of transcription, open chromatin, and enhancers. Our findings implicate TOP2A cleavage as a broad DNA damage mechanism in oncogenic translocations as well as a functional role of TOP2A cleavage in regulating transcription elongation and gene activation.
© 2017 Yu et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
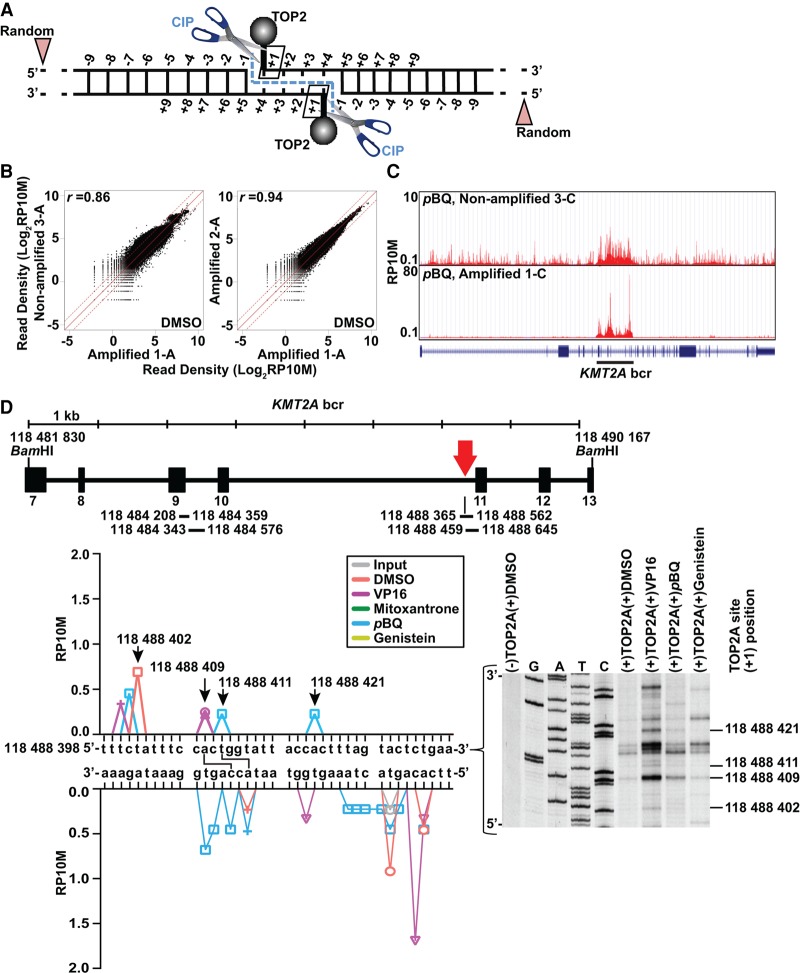
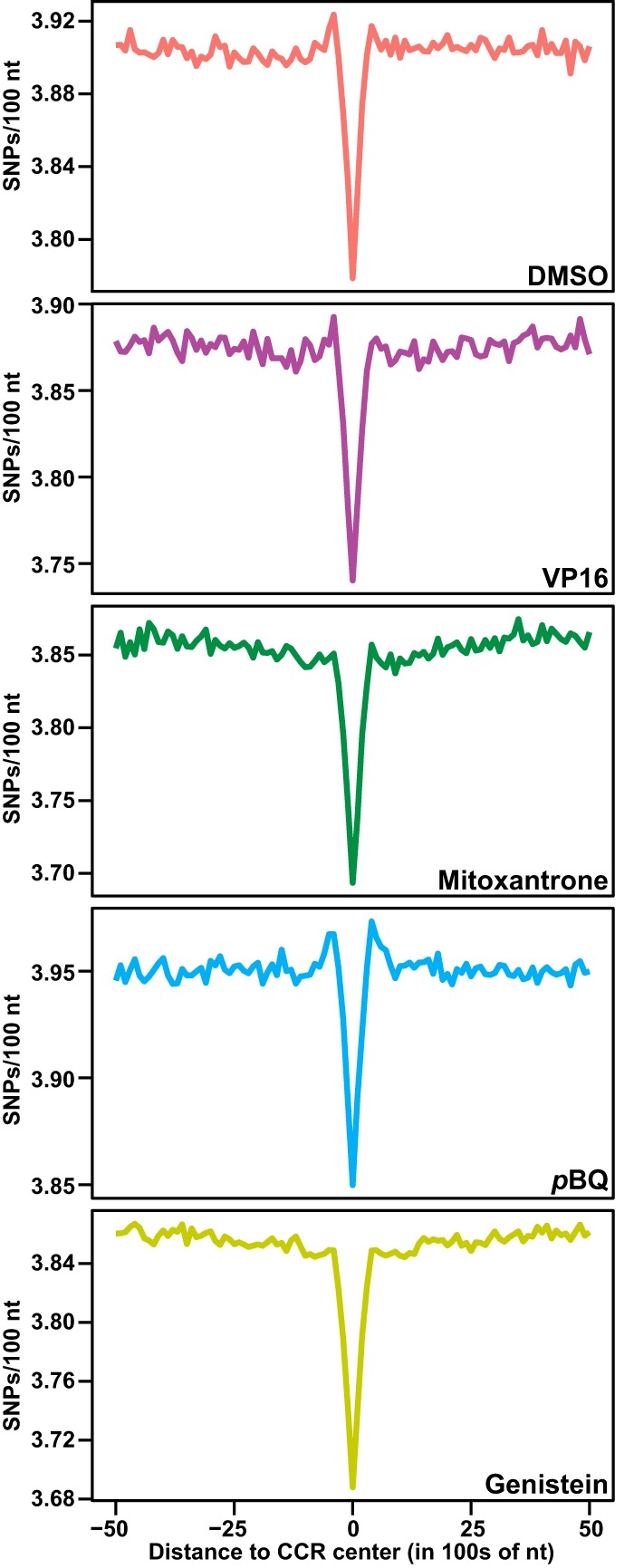
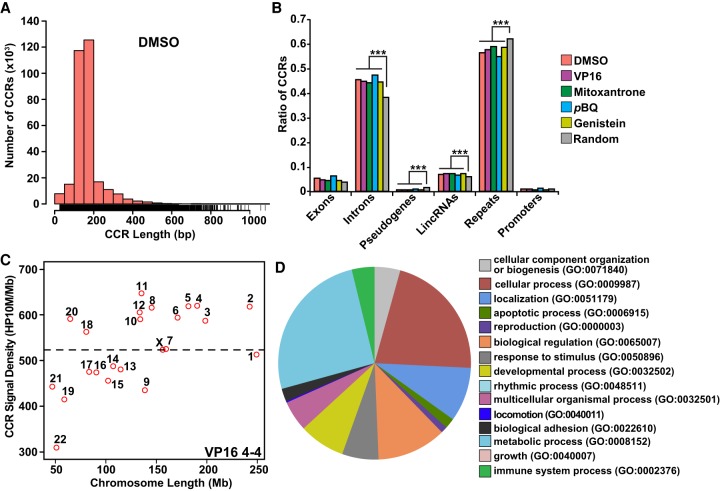
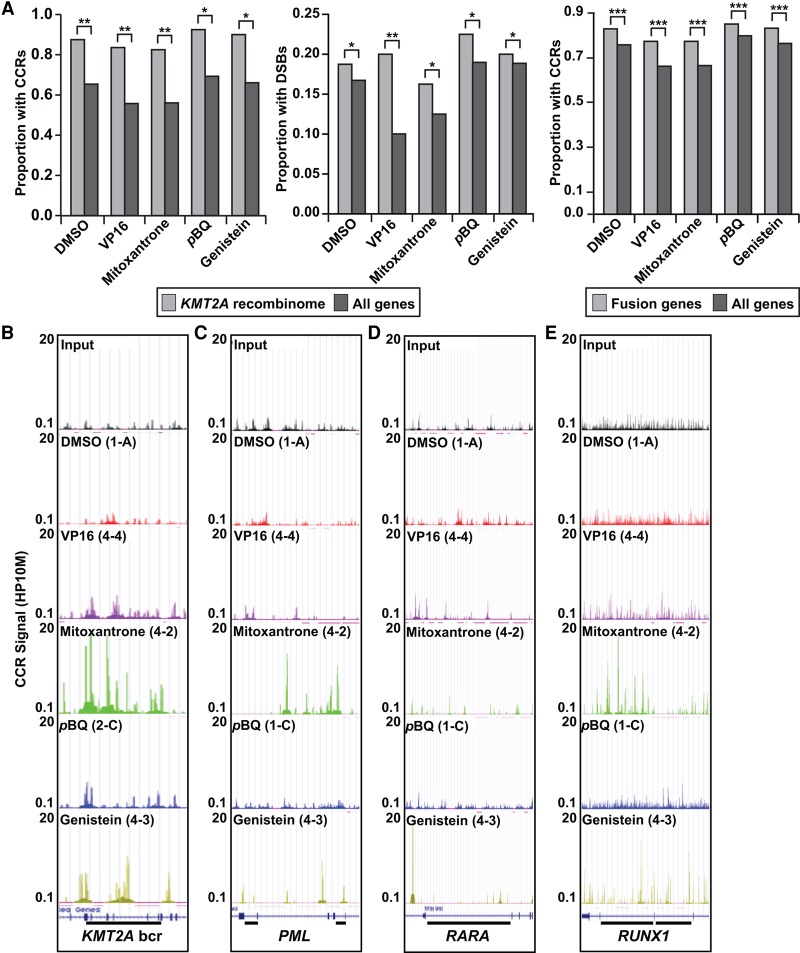
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous