

Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout
- PMID: 28386072
- PMCID: PMC5429603
- DOI: 10.1038/s41598-017-00706-7
Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout
Abstract
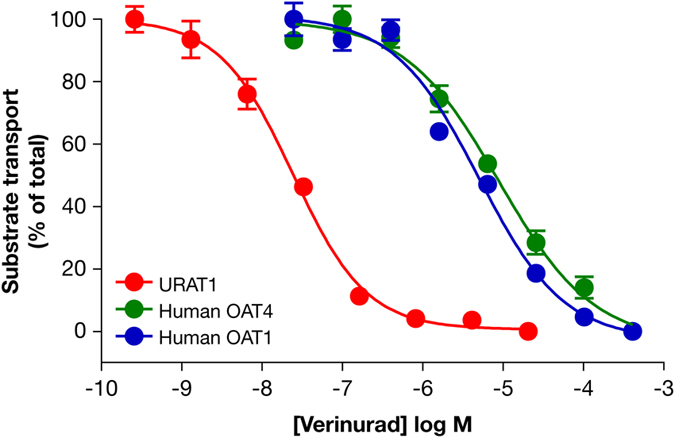
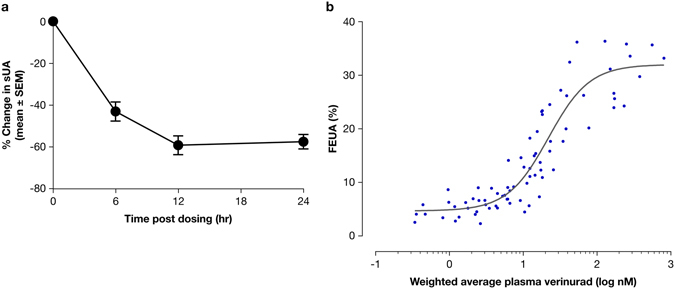
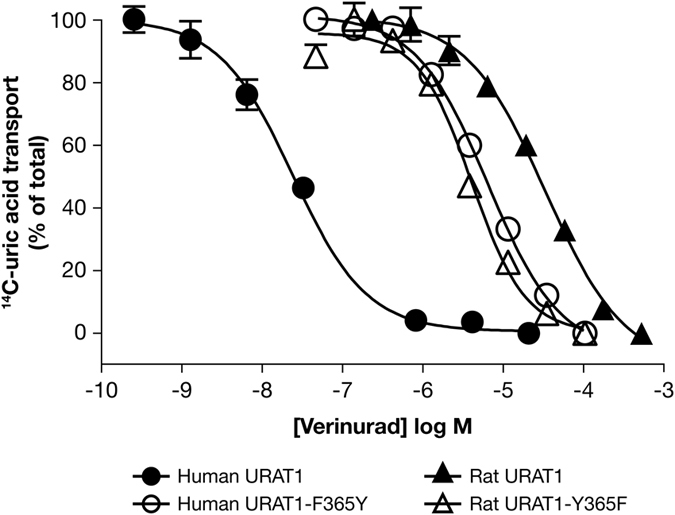
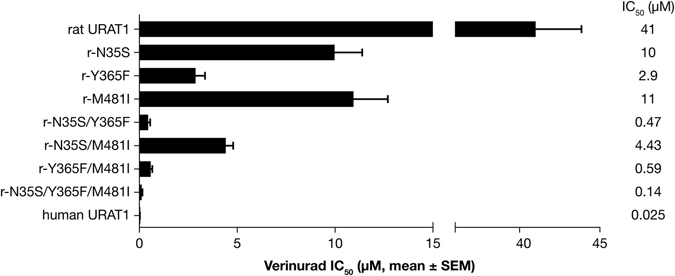
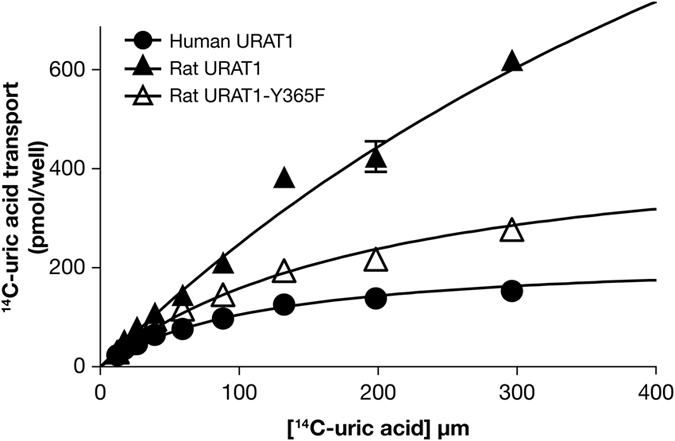
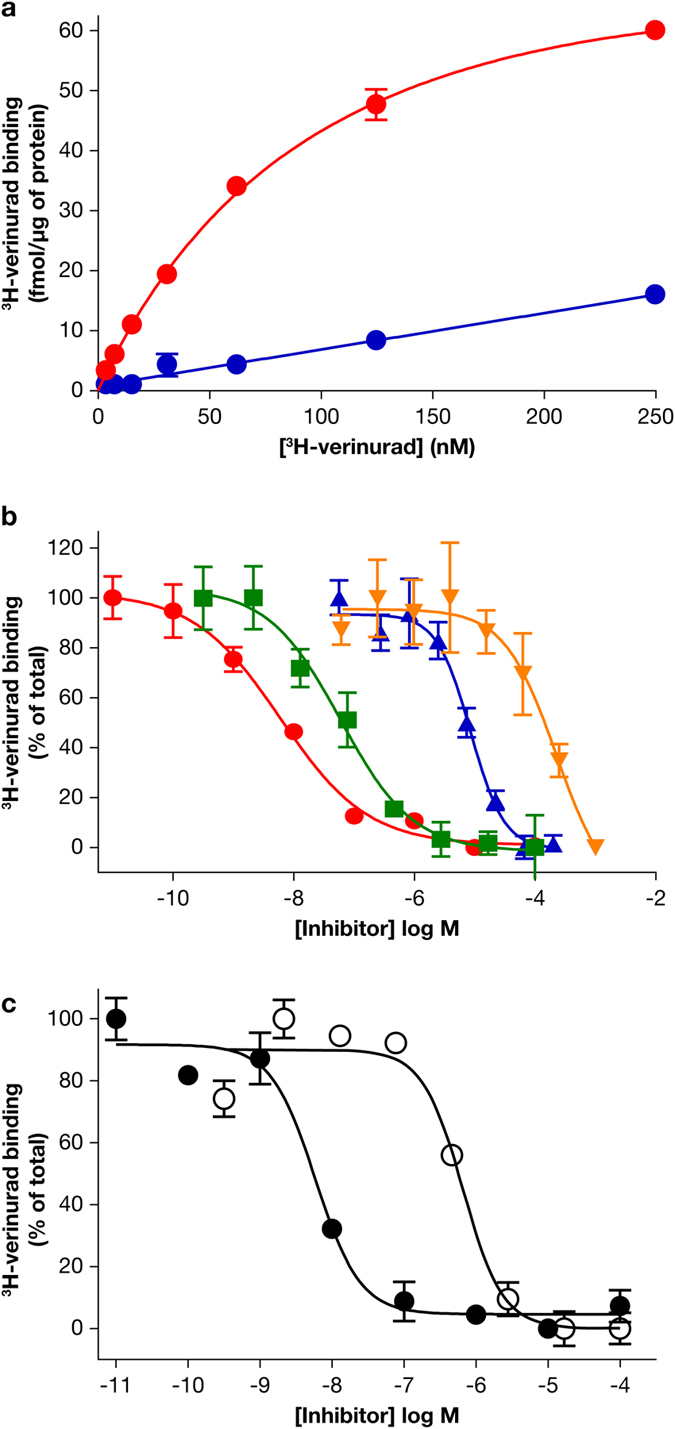
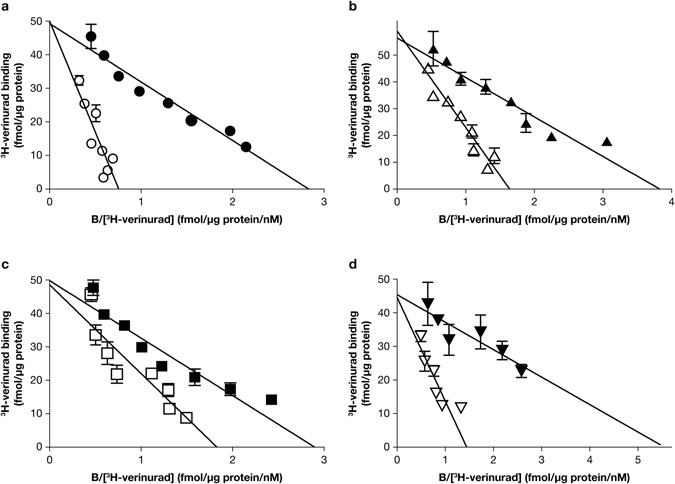
Gout is caused by elevated serum urate levels, which can be treated using inhibitors of the uric acid transporter, URAT1. Here, we characterize verinurad (RDEA3170), which is currently under evaluation for gout therapy. Verinurad specifically inhibits URAT1 with a potency of 25 nM. High affinity inhibition of uric acid transport requires URAT1 residues Cys-32, Ser-35, Phe-365 and Ile-481. Unlike other available uricosuric agents, the requirement for Cys-32 is unique to verinurad. Two of these residues, Ser-35 and Phe-365, are also important for urate transport kinetics. A URAT1 binding assay using radiolabeled verinurad revealed that distinct URAT1 inhibitors benzbromarone, sulfinpyrazone and probenecid all inhibit verinurad binding via a competitive mechanism. However, mutations made within the predicted transporter substrate channel differentially altered the potency for individual URAT1 inhibitors. Overall, our results suggest that URAT1 inhibitors bind to a common site in the core of the transporter and sterically hinder the transit of uric acid through the substrate channel, albeit with vastly different potencies and with differential interactions with specific URAT1 amino acids.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
