

Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration
- PMID: 28387218
- PMCID: PMC5385568
- DOI: 10.1038/ncomms14819
Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration
Abstract
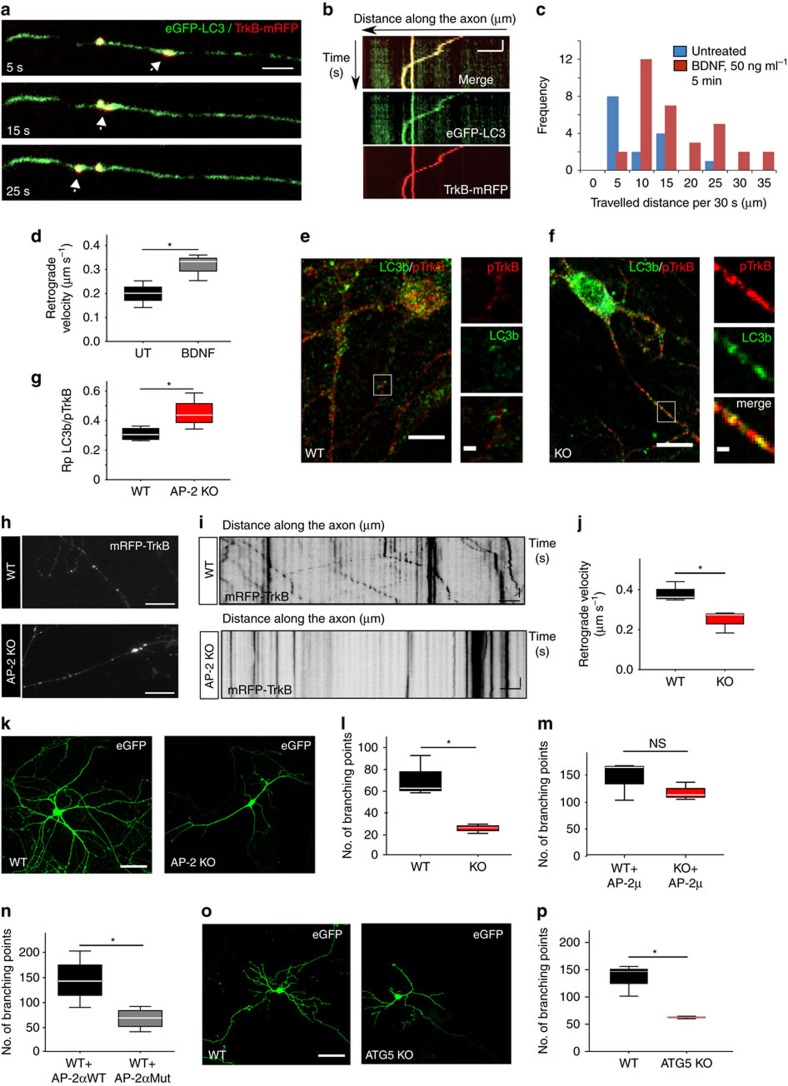
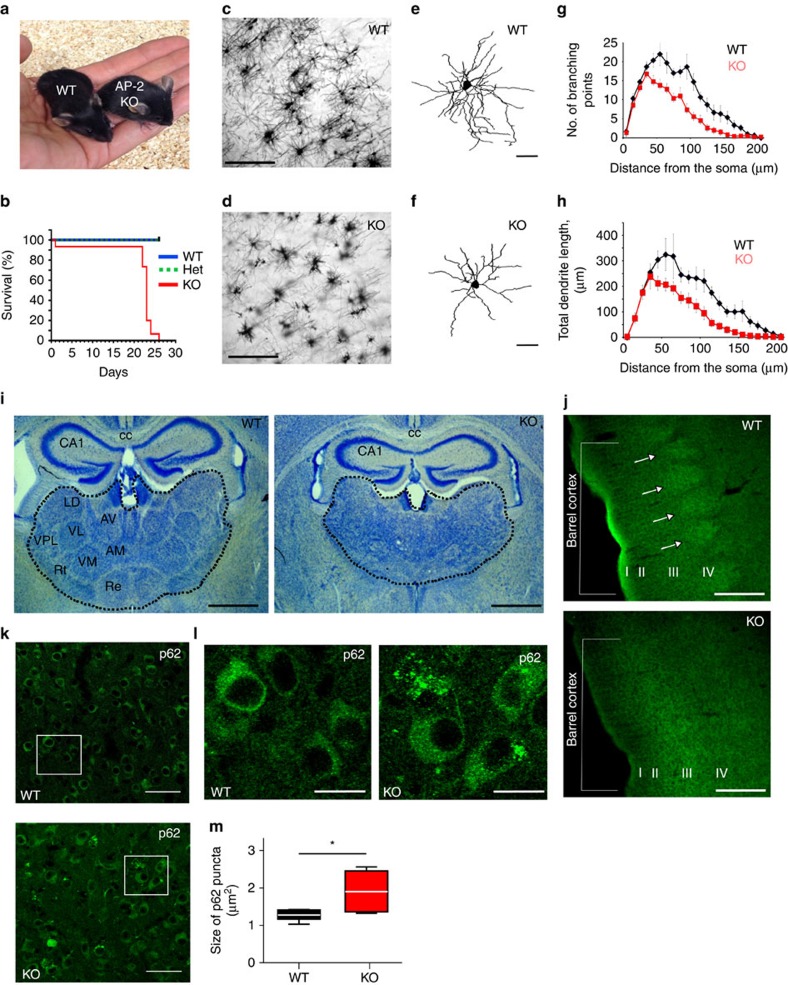
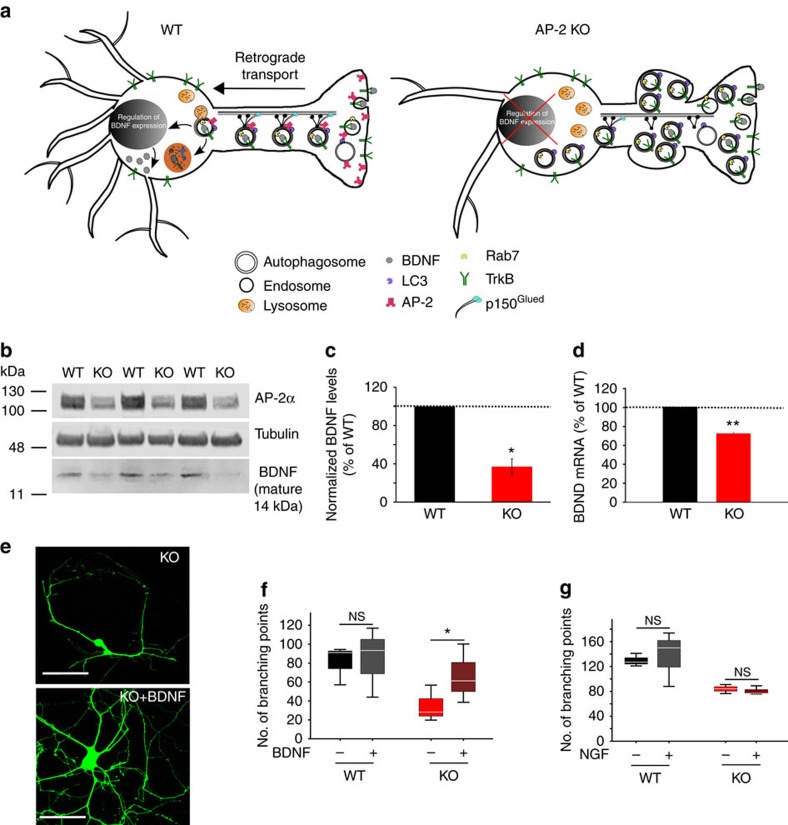
Autophagosomes primarily mediate turnover of cytoplasmic proteins or organelles to provide nutrients and eliminate damaged proteins. In neurons, autophagosomes form in distal axons and are trafficked retrogradely to fuse with lysosomes in the soma. Although defective neuronal autophagy is associated with neurodegeneration, the function of neuronal autophagosomes remains incompletely understood. We show that in neurons, autophagosomes promote neuronal complexity and prevent neurodegeneration in vivo via retrograde transport of brain-derived neurotrophic factor (BDNF)-activated TrkB receptors. p150Glued/dynactin-dependent transport of TrkB-containing autophagosomes requires their association with the endocytic adaptor AP-2, an essential protein complex previously thought to function exclusively in clathrin-mediated endocytosis. These data highlight a novel non-canonical function of AP-2 in retrograde transport of BDNF/TrkB-containing autophagosomes in neurons and reveal a causative link between autophagy and BDNF/TrkB signalling.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
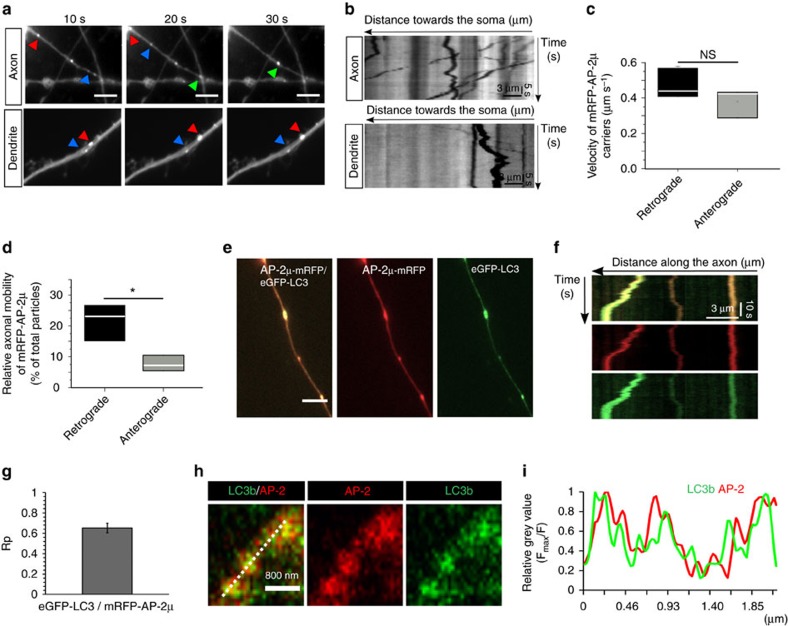
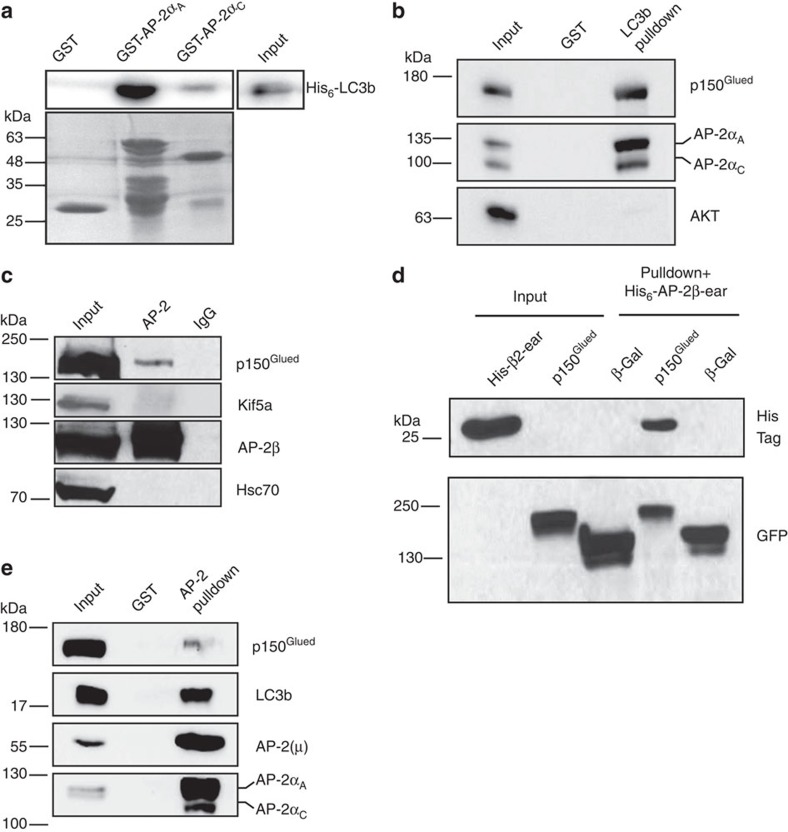
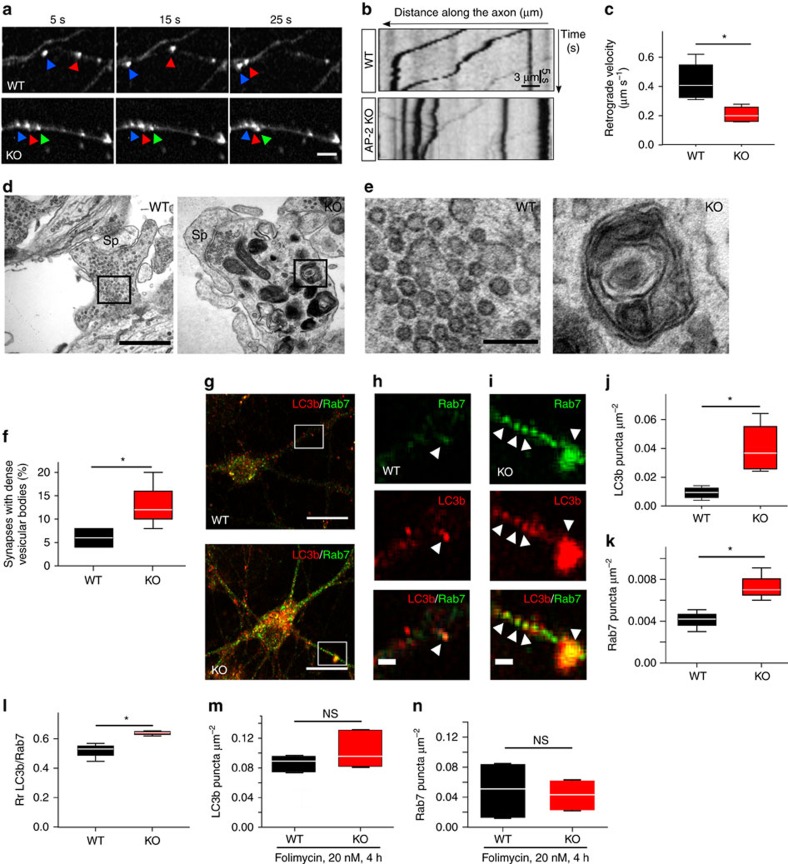
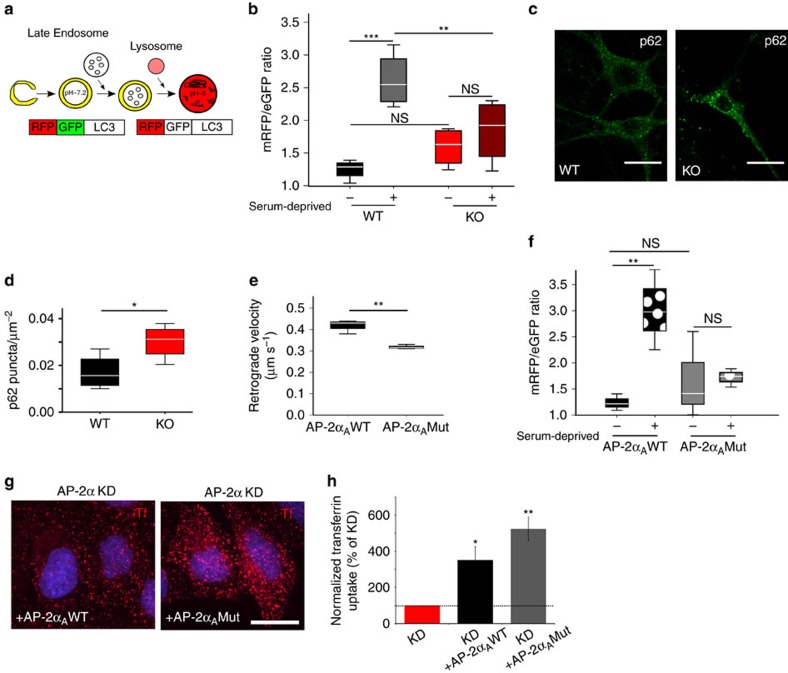
References
-
- Xie Z. & Klionsky D. J. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 (2007). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
