

An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells
- PMID: 28388416
- PMCID: PMC5616215
- DOI: 10.1016/j.cell.2017.03.022
An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells
Abstract
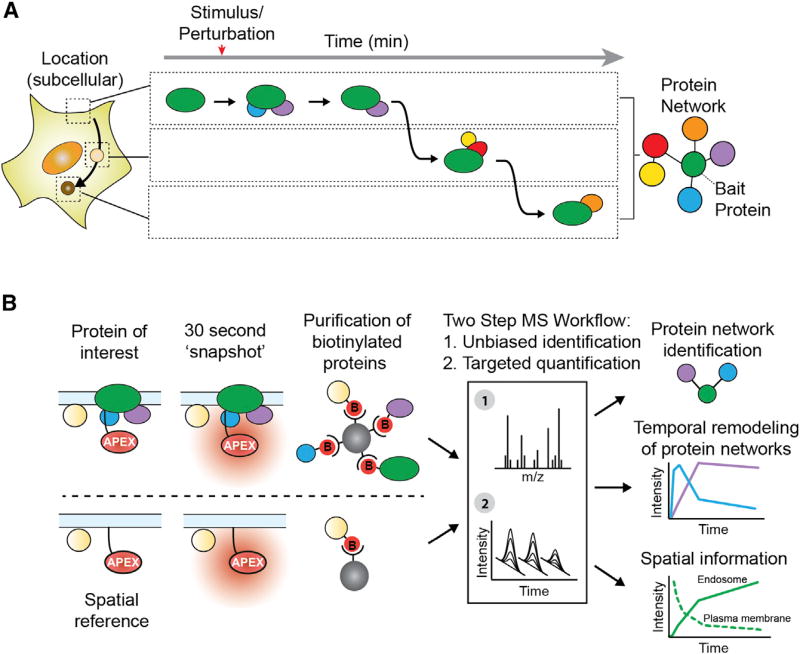
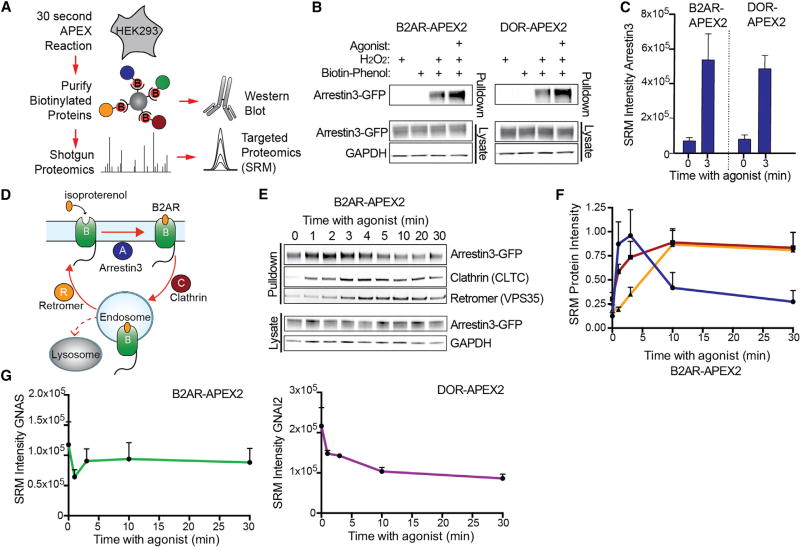
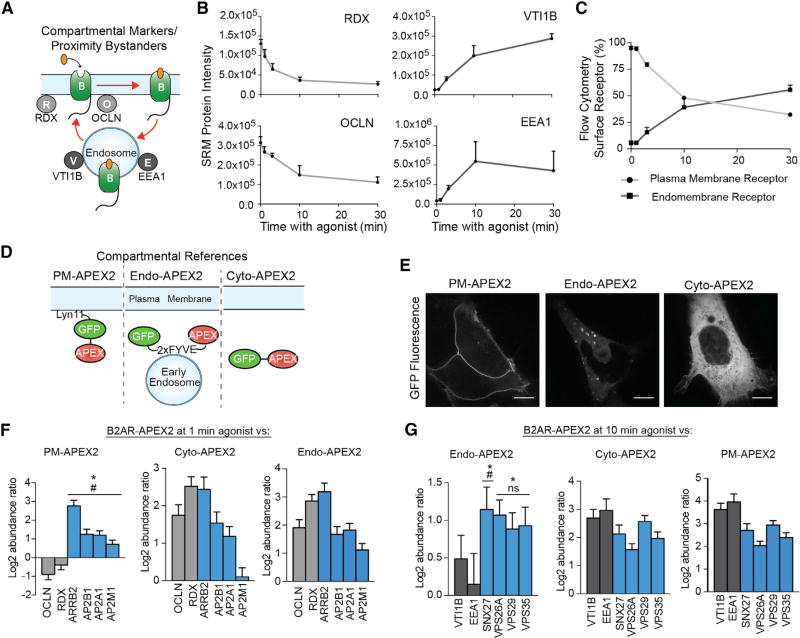
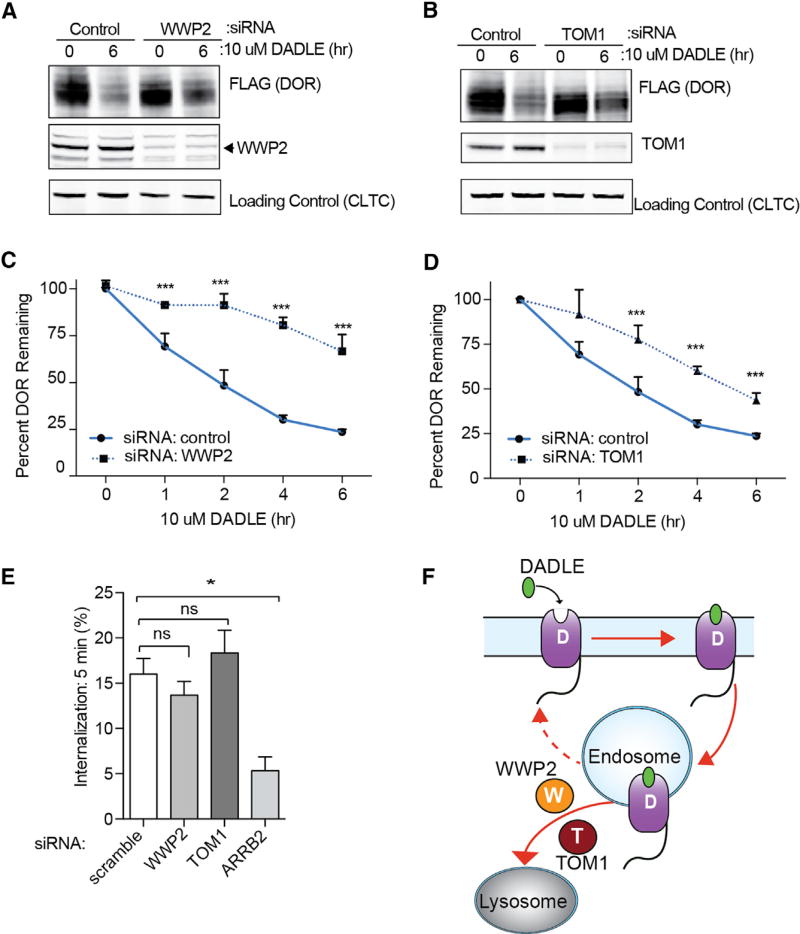
Cells operate through protein interaction networks organized in space and time. Here, we describe an approach to resolve both dimensions simultaneously by using proximity labeling mediated by engineered ascorbic acid peroxidase (APEX). APEX has been used to capture entire organelle proteomes with high temporal resolution, but its breadth of labeling is generally thought to preclude the higher spatial resolution necessary to interrogate specific protein networks. We provide a solution to this problem by combining quantitative proteomics with a system of spatial references. As proof of principle, we apply this approach to interrogate proteins engaged by G-protein-coupled receptors as they dynamically signal and traffic in response to ligand-induced activation. The method resolves known binding partners, as well as previously unidentified network components. Validating its utility as a discovery pipeline, we establish that two of these proteins promote ubiquitin-linked receptor downregulation after prolonged activation.
Keywords: APEX; GPCR; adrenergic receptor; mass spectrometry; opioid receptor; proximity labeling.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures

References
-
- Bernatchez PN, Acevedo L, Fernandez-Hernando C, Murata T, Chalouni C, Kim J, Erdjument-Bromage H, Shah V, Gratton J-P, McNally EM, et al. Myoferlin regulates vascular endothelial growth factor receptor-2 stability and function. J. Biol. Chem. 2007;282:30745–30753. - PubMed
-
- Bisson N, James DA, Ivosev G, Tate SA, Bonner R, Taylor L, Pawson T. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 2011;29:653–658. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 DA010154/DA/NIDA NIH HHS/United States
- P01 HL089707/HL/NHLBI NIH HHS/United States
- T32 GM007810/GM/NIGMS NIH HHS/United States
- R37 DA010711/DA/NIDA NIH HHS/United States
- R01 AI120694/AI/NIAID NIH HHS/United States
- F32 DA038947/DA/NIDA NIH HHS/United States
- R29 DA010711/DA/NIDA NIH HHS/United States
- R01 CA186568/CA/NCI NIH HHS/United States
- P50 GM082250/GM/NIGMS NIH HHS/United States
- U19 AI118610/AI/NIAID NIH HHS/United States
- P30 AI027763/AI/NIAID NIH HHS/United States
- U19 AI106754/AI/NIAID NIH HHS/United States
- P01 AI063302/AI/NIAID NIH HHS/United States
- R01 DA012864/DA/NIDA NIH HHS/United States
- R01 DA010711/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
