

RNA-binding proteins with prion-like domains in health and disease
- PMID: 28389532
- PMCID: PMC5639257
- DOI: 10.1042/BCJ20160499
RNA-binding proteins with prion-like domains in health and disease
Abstract
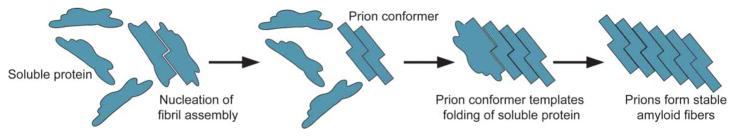
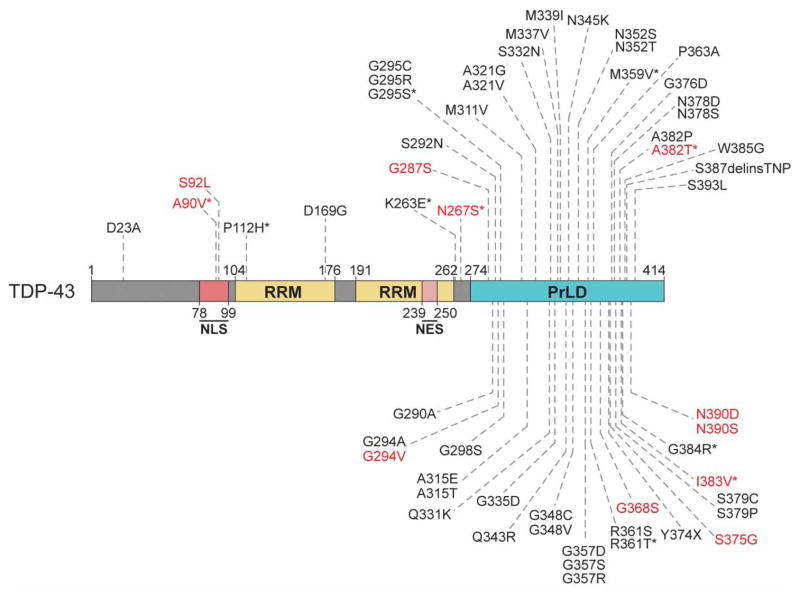
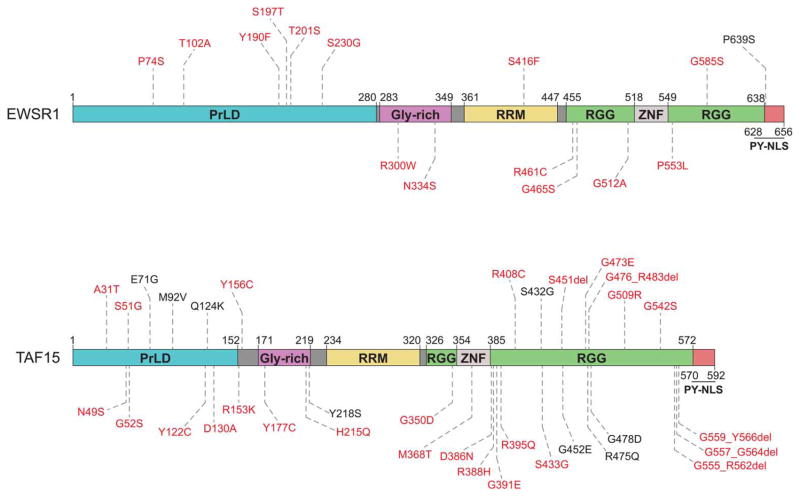
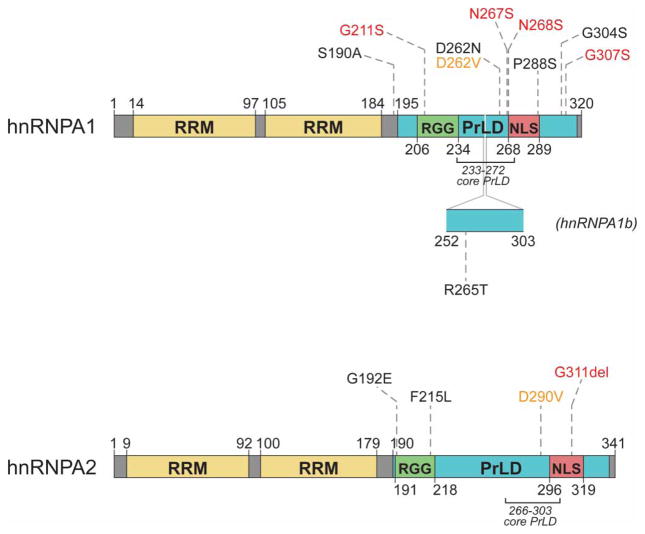
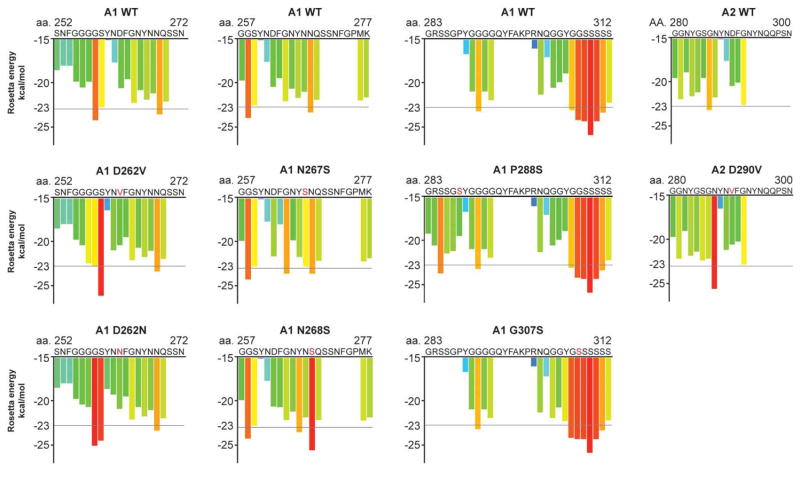
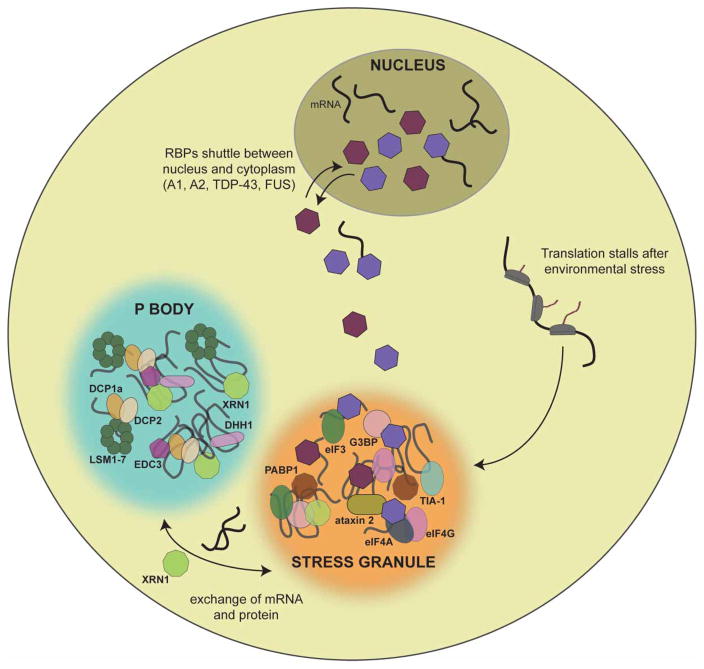
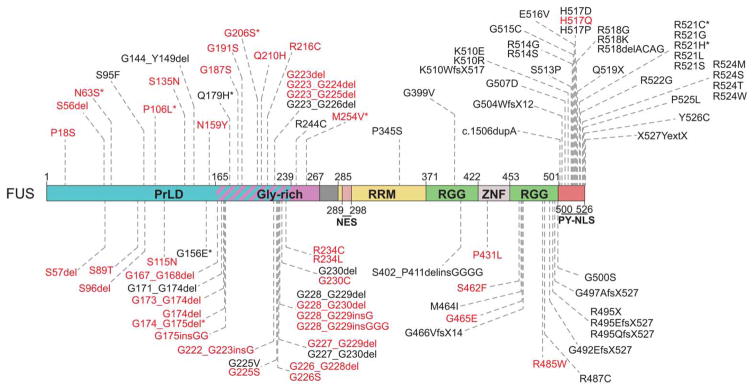
Approximately 70 human RNA-binding proteins (RBPs) contain a prion-like domain (PrLD). PrLDs are low-complexity domains that possess a similar amino acid composition to prion domains in yeast, which enable several proteins, including Sup35 and Rnq1, to form infectious conformers, termed prions. In humans, PrLDs contribute to RBP function and enable RBPs to undergo liquid-liquid phase transitions that underlie the biogenesis of various membraneless organelles. However, this activity appears to render RBPs prone to misfolding and aggregation connected to neurodegenerative disease. Indeed, numerous RBPs with PrLDs, including TDP-43 (transactivation response element DNA-binding protein 43), FUS (fused in sarcoma), TAF15 (TATA-binding protein-associated factor 15), EWSR1 (Ewing sarcoma breakpoint region 1), and heterogeneous nuclear ribonucleoproteins A1 and A2 (hnRNPA1 and hnRNPA2), have now been connected via pathology and genetics to the etiology of several neurodegenerative diseases, including amyotrophic lateral sclerosis, frontotemporal dementia, and multisystem proteinopathy. Here, we review the physiological and pathological roles of the most prominent RBPs with PrLDs. We also highlight the potential of protein disaggregases, including Hsp104, as a therapeutic strategy to combat the aberrant phase transitions of RBPs with PrLDs that likely underpin neurodegeneration.
Keywords: RNA-binding proteins; disaggregase; neurodegeneration; phase separation; prion-like domain.
© 2017 The Author(s); published by Portland Press Limited on behalf of the Biochemical Society.
Conflict of interest statement
The Authors declare that there are no competing interests associated with the manuscript.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- T32 AG000255/AG/NIA NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- R01 GM099836/GM/NIGMS NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- R21 NS090205/NS/NINDS NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- F31 NS087676/NS/NINDS NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases