

Artemisinin protects PC12 cells against β-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway
- PMID: 28391183
- PMCID: PMC5385605
- DOI: 10.1016/j.redox.2017.04.003
Artemisinin protects PC12 cells against β-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway
Abstract
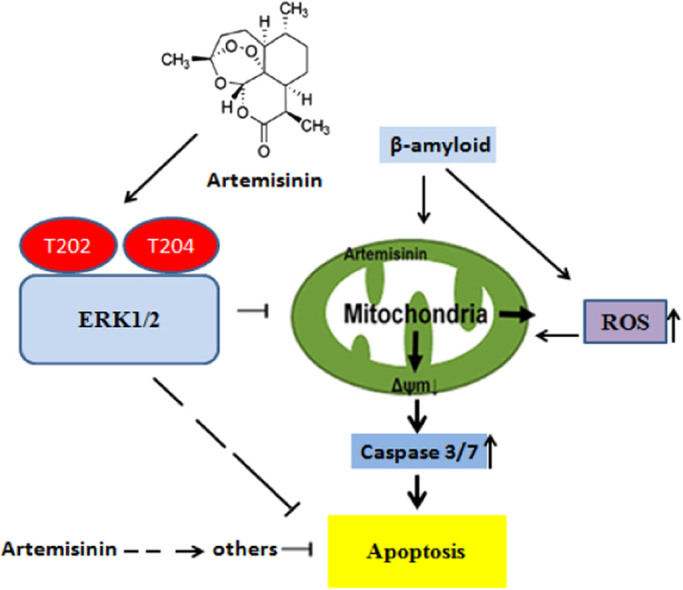
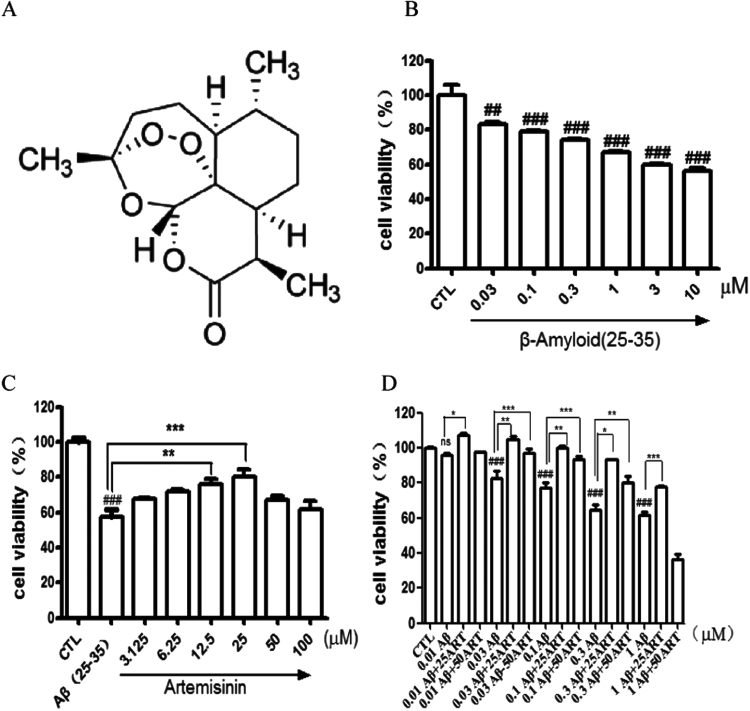
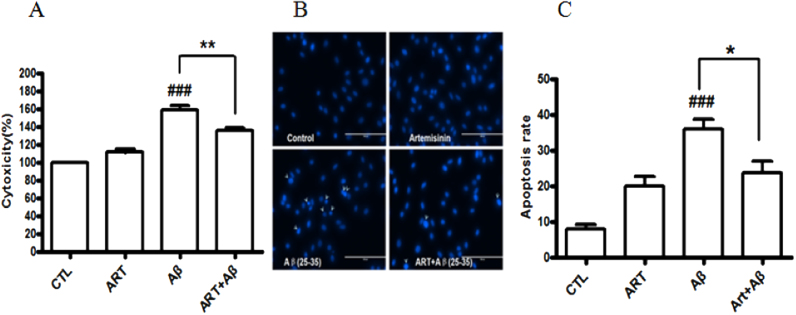
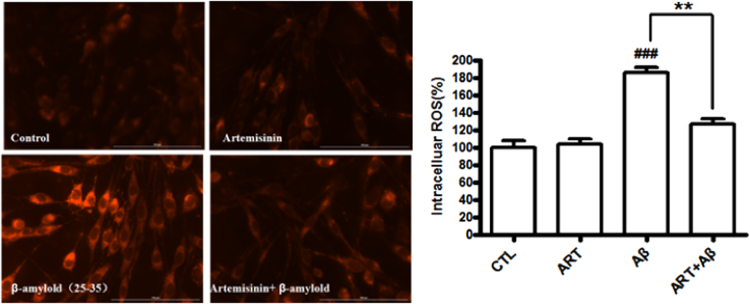
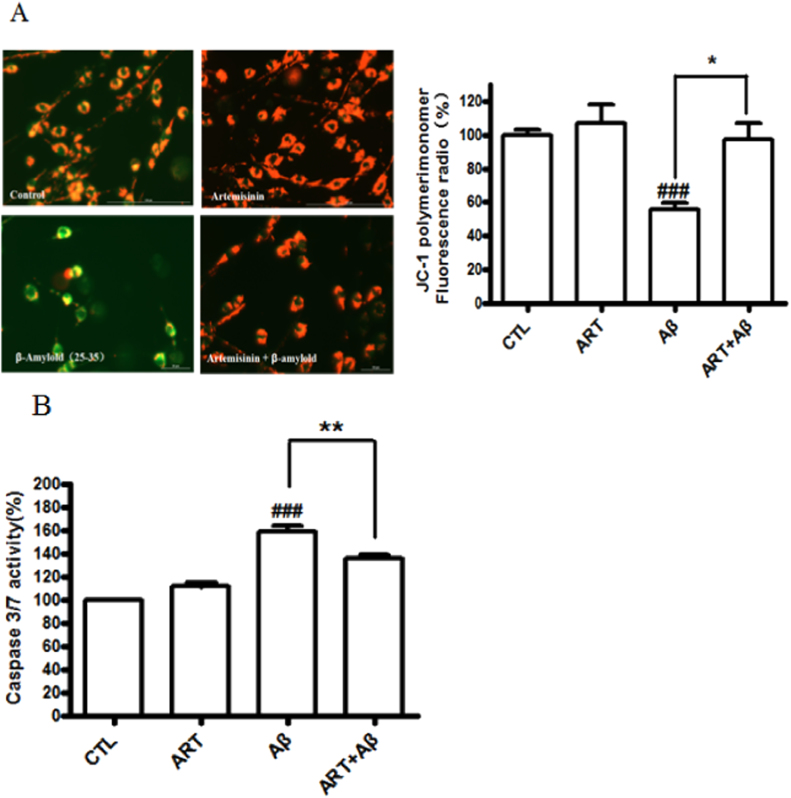
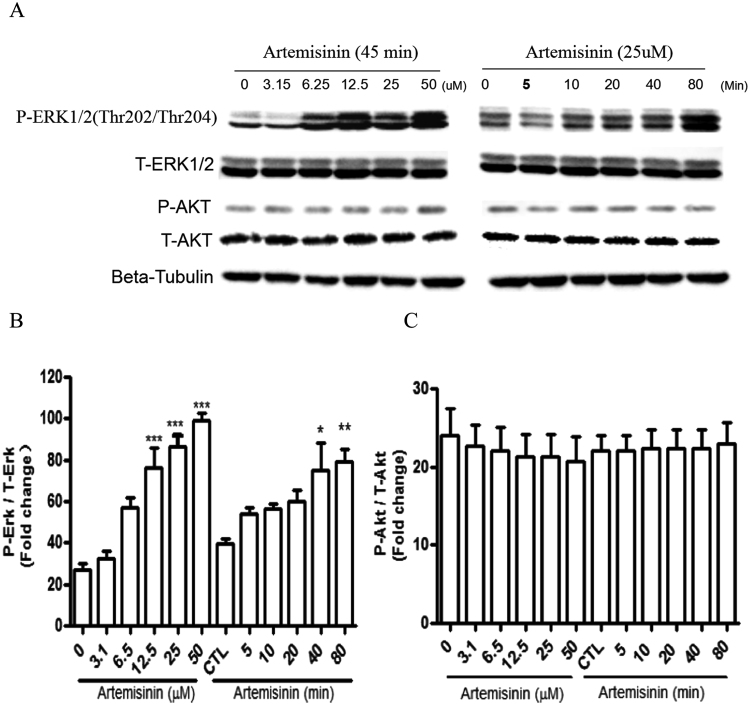
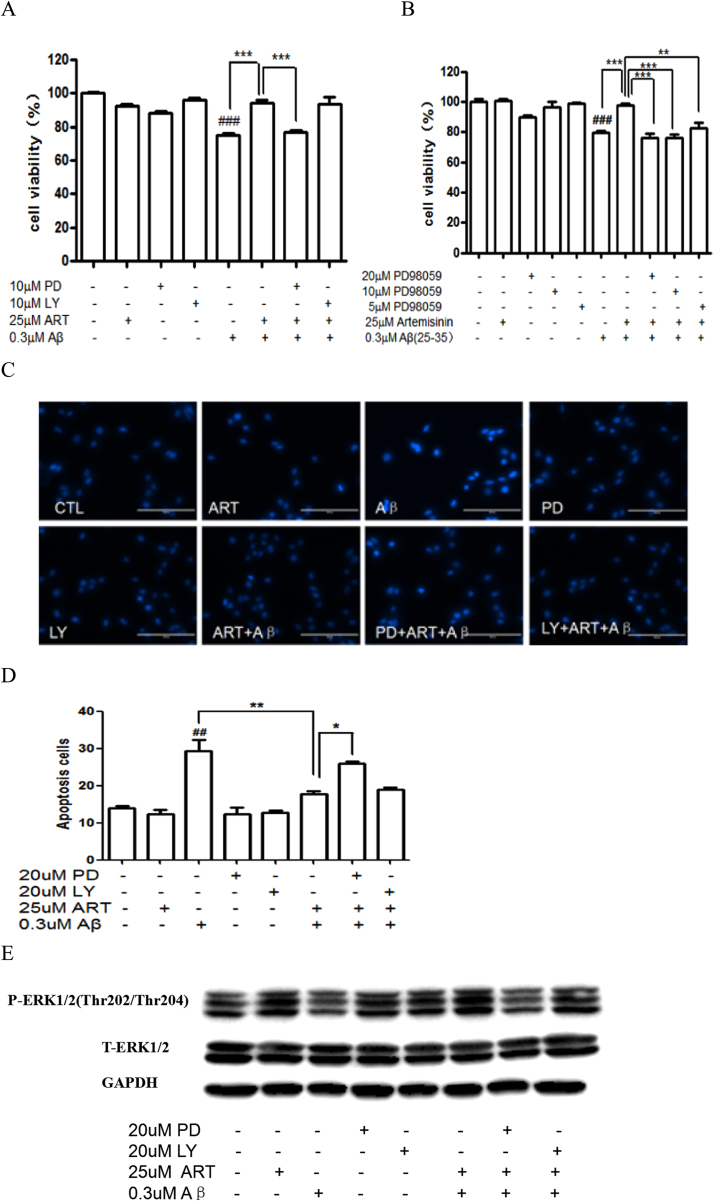
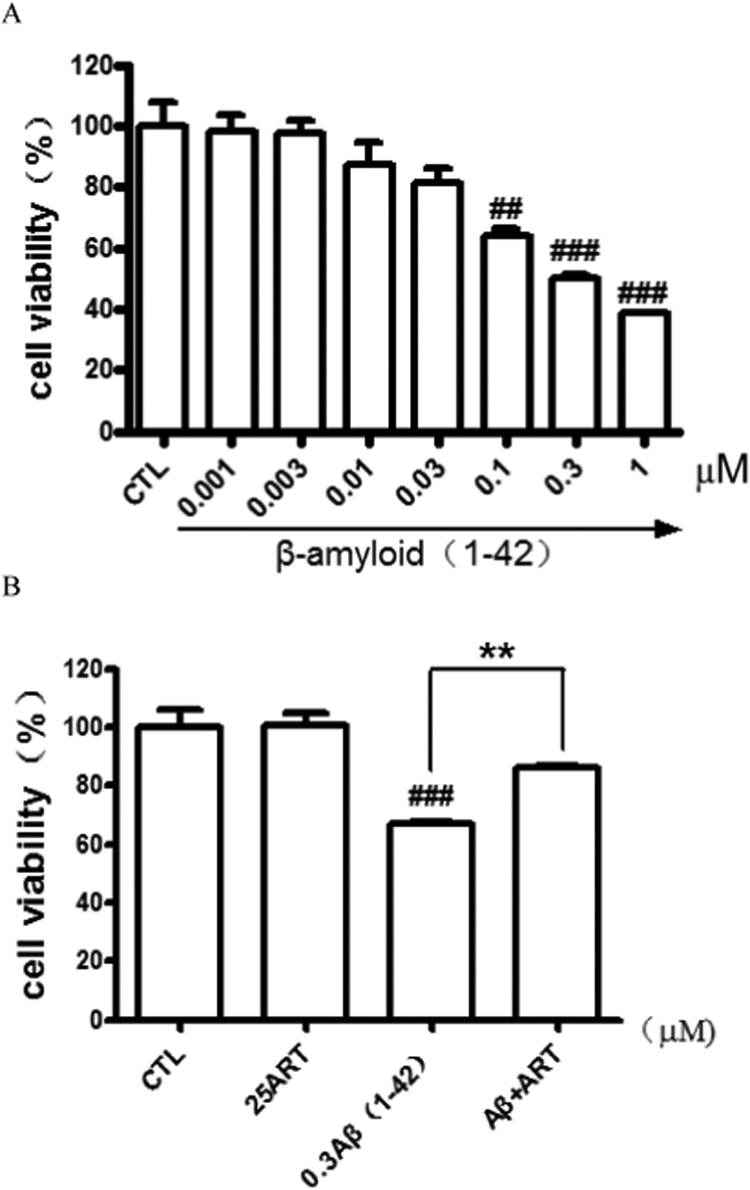
Accumulating evidence displays that an abnormal deposition of amyloid beta-peptide (Aβ) is the primary cause of the pathogenesis of Alzheimer's disease (AD). And therefore the elimination of Aβ is regarded as an important strategy for AD treatment. The discovery of drug candidates using culture neuronal cells against Aβ peptide toxicity is believed to be an effective approach to develop drug for the treatment of AD patients. We have previously showed that artemisinin, a FDA-approved anti-malaria drug, has neuroprotective effects recently. In the present study, we aimed to investigate the effects and potential mechanism of artemisinin in protecting neuronal PC12 cells from toxicity of β amyloid peptide. Our studies revealed that artemisinin, in clinical relevant concentration, protected and rescued PC12 cells from Aβ25-35-induced cell death. Further study showed that artemisinin significantly ameliorated cell death due to Aβ25-35 insult by restoring abnormal changes in nuclear morphology, lactate dehydrogenase, intracellular ROS, mitochondrial membrane potential and activity of apoptotic caspase. Western blotting analysis demonstrated that artemisinin activated extracellular regulated kinase ERK1/2 but not Akt survival signaling. Consistent with the role of ERK1/2, preincubation of cells with ERK1/2 pathway inhibitor PD98059 blocked the effect of artemisinin while PI3K inhibitor LY294002 has no effect. Moreover, Aβ1-42 also caused cells death of PC12 cells while artemisinin suppressed Aβ1-42 cytotoxicity in PC12 cells. Taken together, these results, at the first time, suggest that artemisinin is a potential protectant against β amyloid insult through activation of the ERK1/2 pathway. Our finding provides a potential application of artemisinin in prevention and treatment of AD.
Keywords: Alzheimer's disease; Artemisinin; Aβ25–35; ERK1/2; PC12 cells.
Copyright © 2017 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Vila M., Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003;4(5):365–375. - PubMed
-
- Forman M.S., Trojanowski J.Q., Lee V.M. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004;10(10):1055–1063. - PubMed
-
- Taylor J.P., Hardy J., Fischbeck K.H. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991–1995. - PubMed
-
- Colucci L. Alzheimer's disease costs: what we know and what we should take into account. J. Alzheimers Dis. 2014;42(4):1311–1324. - PubMed
-
- Selkoe D.J. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789–791. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
