

Generation of induced Pluripotent Stem Cells as disease modelling of NLSDM
- PMID: 28391974
- PMCID: PMC5434246
- DOI: 10.1016/j.ymgme.2017.03.009
Generation of induced Pluripotent Stem Cells as disease modelling of NLSDM
Abstract
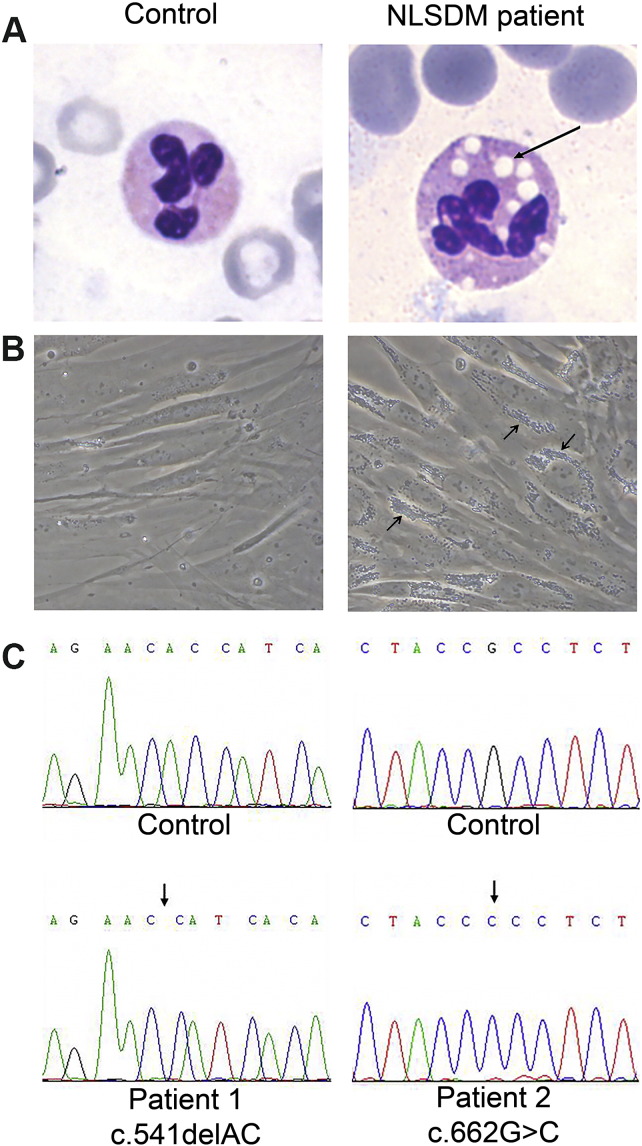
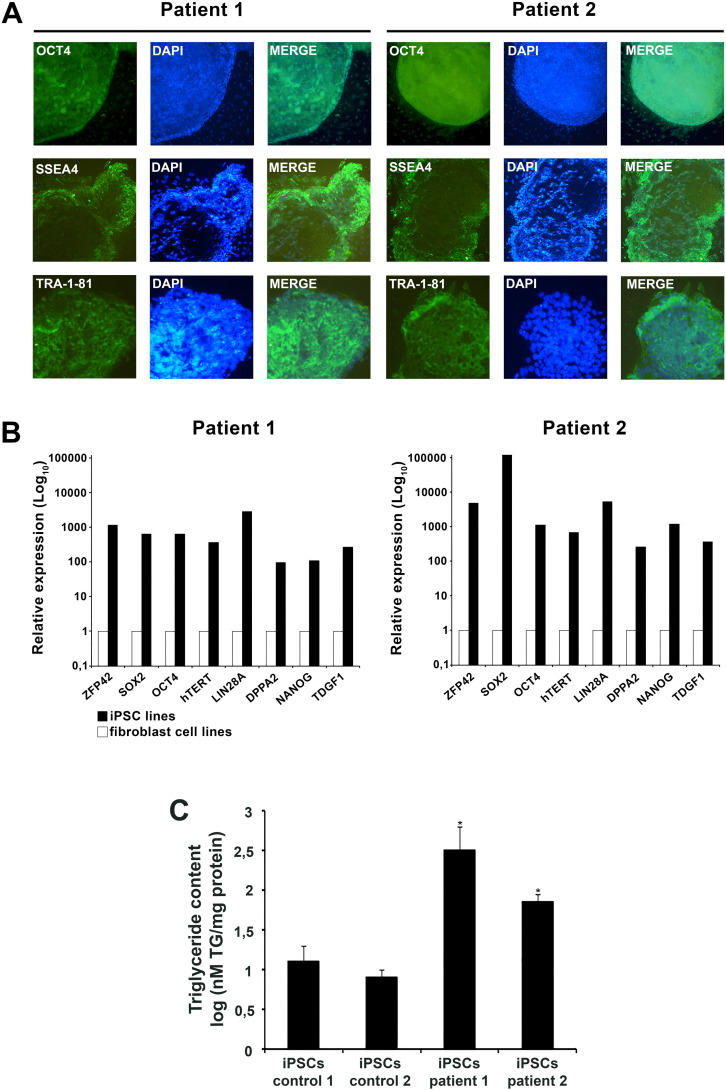
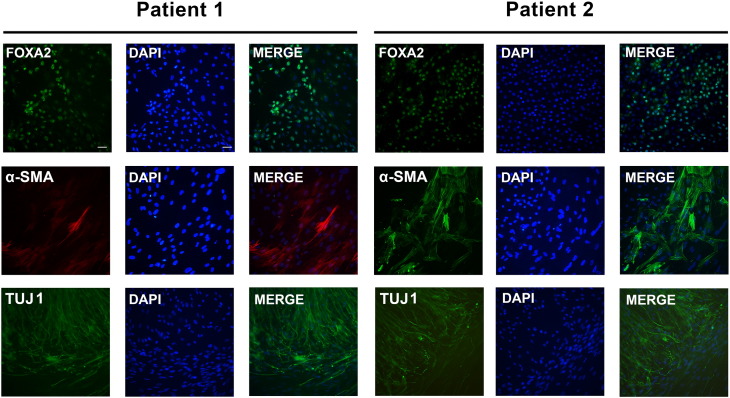
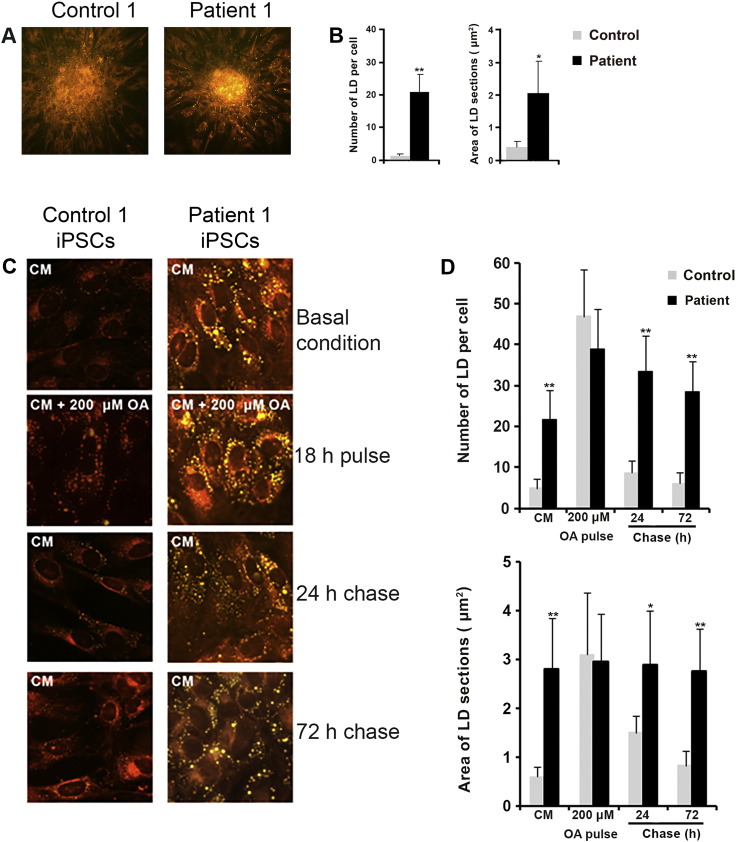
Neutral Lipid Storage Disease with Myopathy (NLSDM) is a rare defect of triacylglycerol metabolism, characterized by the abnormal storage of neutral lipid in organelles known as lipid droplets (LDs). The main clinical features are progressive myopathy and cardiomyopathy. The onset of NLSDM is caused by autosomal recessive mutations in the PNPLA2 gene, which encodes adipose triglyceride lipase (ATGL). Despite its name, this enzyme is present in a wide variety of cell types and catalyzes the first step in triacylglycerol lipolysis and the release of fatty acids. Here, we report the derivation of NLSDM-induced pluripotent stem cells (NLSDM-iPSCs) from fibroblasts of two patients carrying different PNPLA2 mutations. The first patient was homozygous for the c.541delAC, while the second was homozygous for the c.662G>C mutation in the PNPLA2 gene. We verified that the two types of NLSDM-iPSCs possessed properties of embryonic-like stem cells and could differentiate into the three germ layers in vitro. Immunofluorescence analysis revealed that iPSCs had an abnormal accumulation of triglycerides in LDs, the hallmark of NLSDM. Furthermore, NLSDM-iPSCs were deficient in long chain fatty acid lipolysis, when subjected to a pulse chase experiment with oleic acid. Collectively, these results demonstrate that NLSDM-iPSCs are a promising in vitro model to investigate disease mechanisms and screen drug compounds for NLSDM, a rare disease with few therapeutic options.
Keywords: Lipid droplets; Lipid metabolism; Myopathy; NLSDM; PNPLA2; iPSCs.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Fischer J., Lefèvre C., Morava E., Mussini J.M., Laforêt P., Negre-Salvayre A., Lathrop M., Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007;39:28–30. - PubMed
-
- Campagna F., Nanni L., Quagliarini F., Pennisi E., Michailidis C., Pierelli F., Bruno C., Casali C., DiMauro S., Arca M. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem. Biophys. Res. Commun. 2008;377:843–846. - PubMed
-
- Tavian D., Missaglia S., Redaelli C., Pennisi E.M., Invernici G., Wessalowski R., Maiwald R., Arca M., Coleman R.A. Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: a strikingly low amount of lipase activity may preserve cardiac function. Hum. Mol. Genet. 2012;21:5318–5328. - PMC - PubMed
-
- Pennisi E.M., Missaglia S., DiMauro S., Bernardi C., Akman H.O., Tavian D. A myopathy with unusual features caused by PNPLA2 gene mutations. Muscle Nerve. 2014;51:609–613. - PubMed
-
- Kaneko K., Kuroda H., Izumi R., Tateyama M., Kato M., Sugimura K., Sakata Y., Ikeda Y., Hirano K., Aoki M. A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: a case report and literature review. Neuromuscul. Disord. 2014;24:634–641. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
