

How Do the Virulence Factors of Shigella Work Together to Cause Disease?
- PMID: 28393050
- PMCID: PMC5364150
- DOI: 10.3389/fcimb.2017.00064
How Do the Virulence Factors of Shigella Work Together to Cause Disease?
Abstract
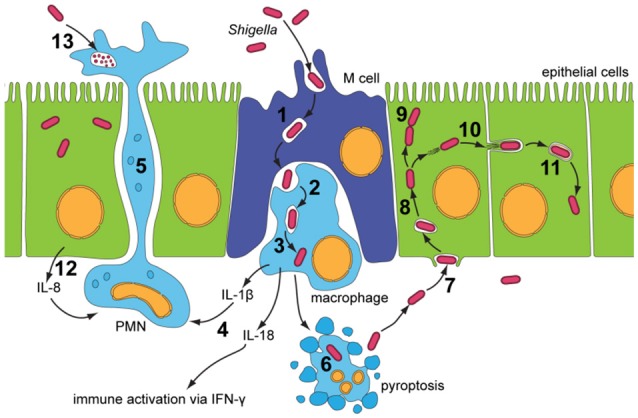
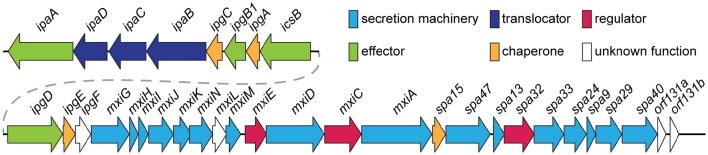
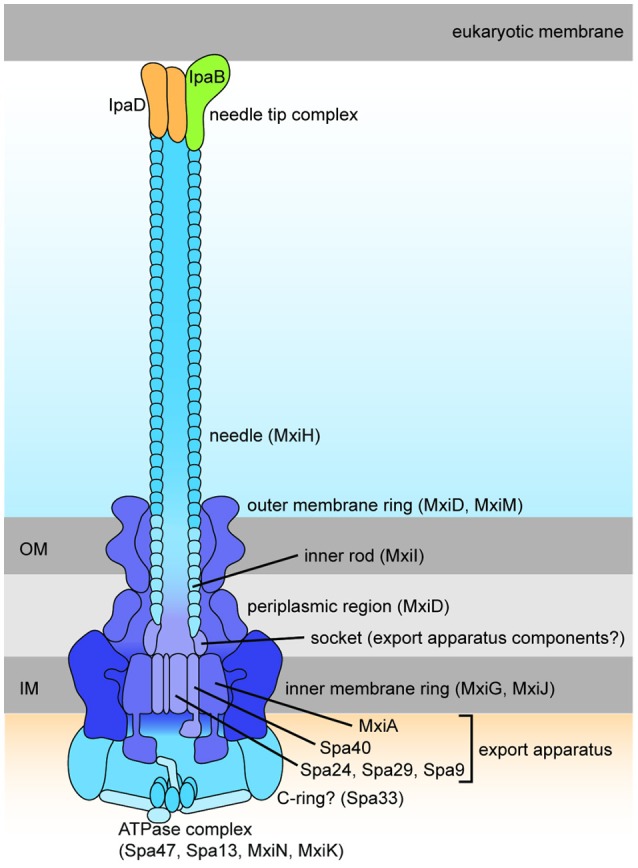
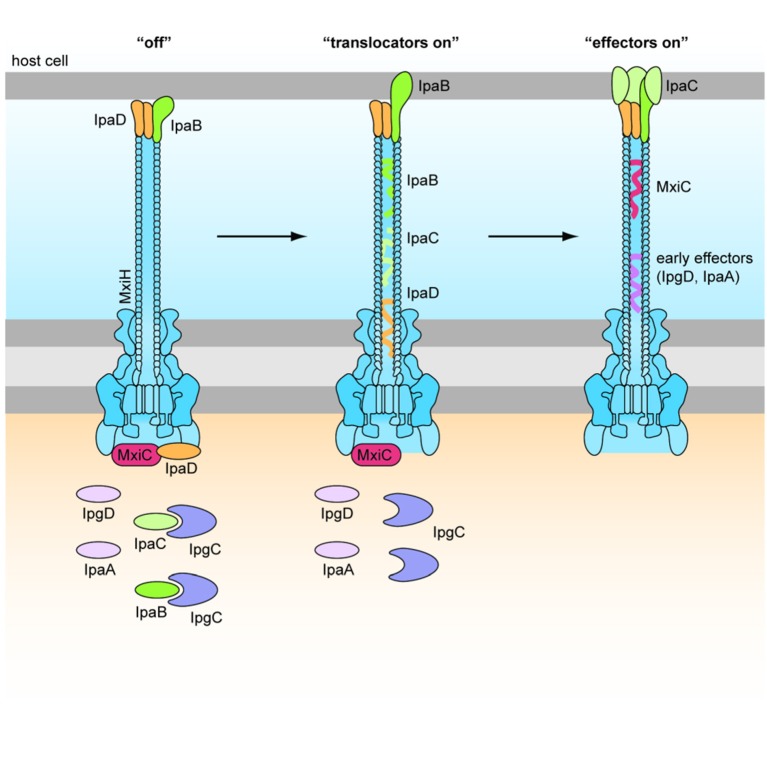
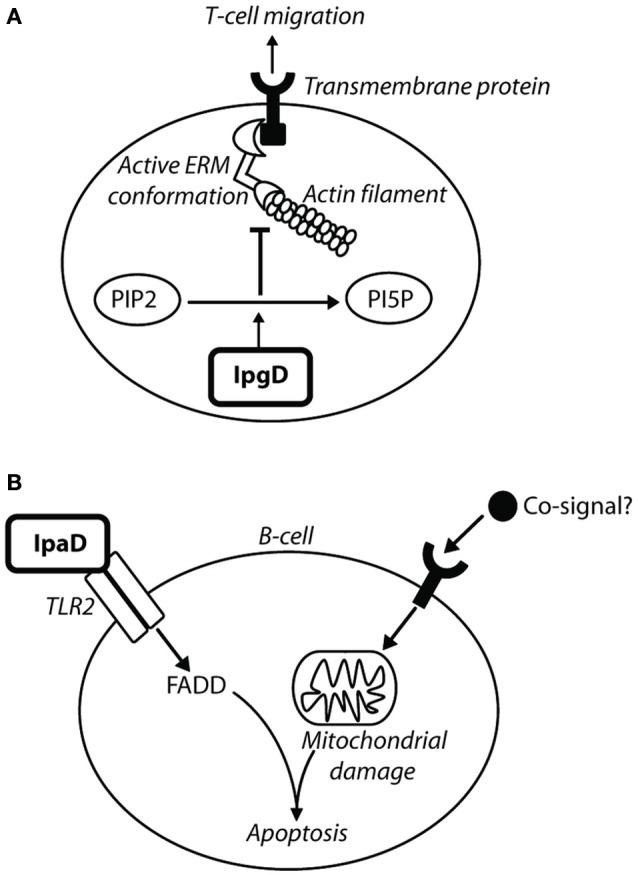
Shigella is the major cause of bacillary dysentery world-wide. It is divided into four species, named S. flexneri, S. sonnei, S. dysenteriae, and S. boydii, which are distinct genomically and in their ability to cause disease. Shigellosis, the clinical presentation of Shigella infection, is characterized by watery diarrhea, abdominal cramps, and fever. Shigella's ability to cause disease has been attributed to virulence factors, which are encoded on chromosomal pathogenicity islands and the virulence plasmid. However, information on these virulence factors is not often brought together to create a detailed picture of infection, and how this translates into shigellosis symptoms. Firstly, Shigella secretes virulence factors that induce severe inflammation and mediate enterotoxic effects on the colon, producing the classic watery diarrhea seen early in infection. Secondly, Shigella injects virulence effectors into epithelial cells via its Type III Secretion System to subvert the host cell structure and function. This allows invasion of epithelial cells, establishing a replicative niche, and causes erratic destruction of the colonic epithelium. Thirdly, Shigella produces effectors to down-regulate inflammation and the innate immune response. This promotes infection and limits the adaptive immune response, causing the host to remain partially susceptible to re-infection. Combinations of these virulence factors may contribute to the different symptoms and infection capabilities of the diverse Shigella species, in addition to distinct transmission patterns. Further investigation of the dominant species causing disease, using whole-genome sequencing and genotyping, will allow comparison and identification of crucial virulence factors and may contribute to the production of a pan-Shigella vaccine.
Keywords: Shigella; Shigellosis; bacterial pathogenesis; type III secretion system; virulence effectors.
Figures
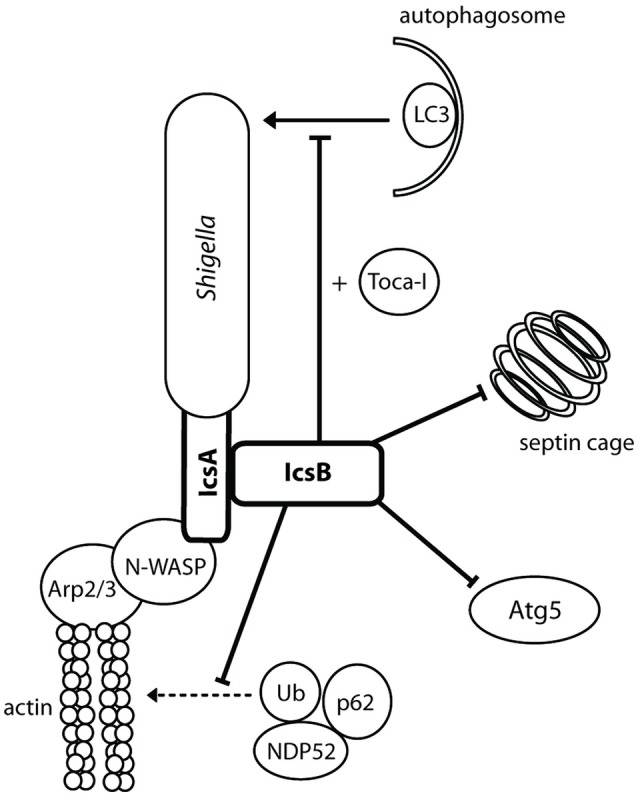
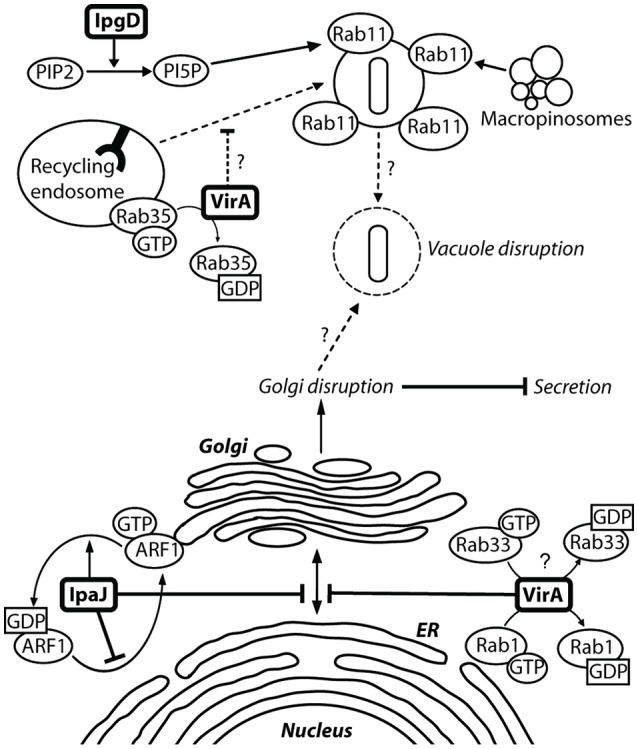
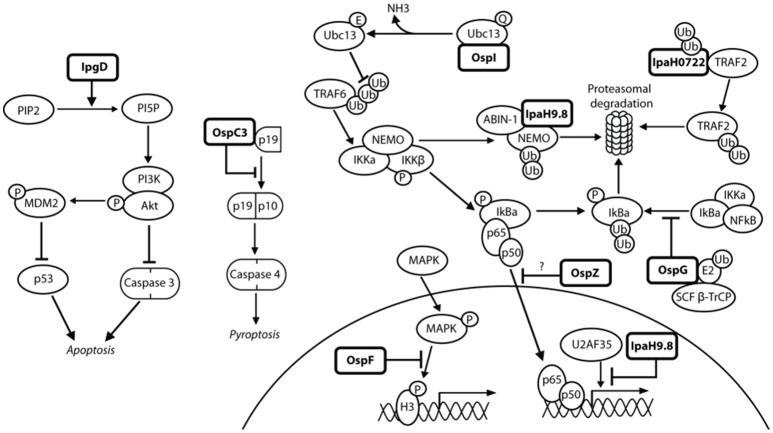
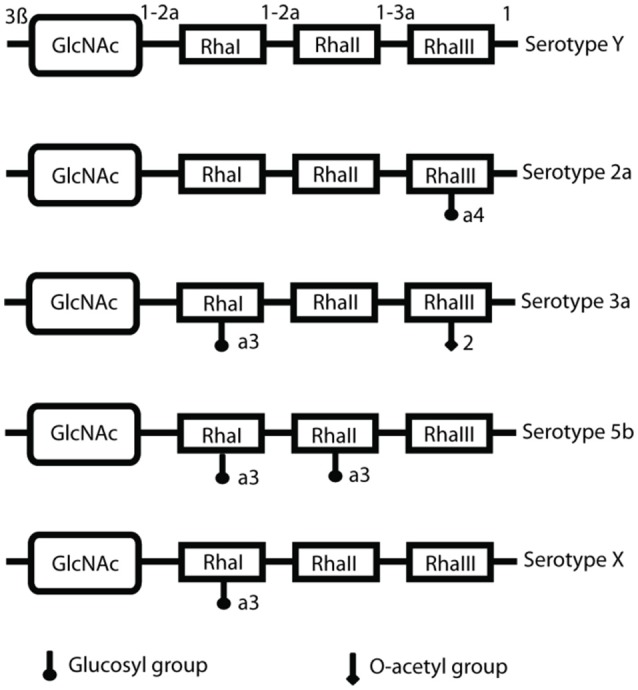
References
-
- Al-Hasani K., Henderson I. R., Sakellaris H., Rajakumar K., Grant T., Nataro J. P., et al. (2000). The sigA gene which is borne on the she pathogenicity island of Shigella flexneri 2a encodes an exported cytopathic protease involved in intestinal fluid accumulation. Infect. Immun. 68, 2457–2463. 10.1128/IAI.68.5.2457-2463.2000 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources