

Genomic Diversity of Type B3 Bacteriophages of Caulobacter crescentus
- PMID: 28393265
- PMCID: PMC5480614
- DOI: 10.1007/s00284-017-1248-4
Genomic Diversity of Type B3 Bacteriophages of Caulobacter crescentus
Abstract
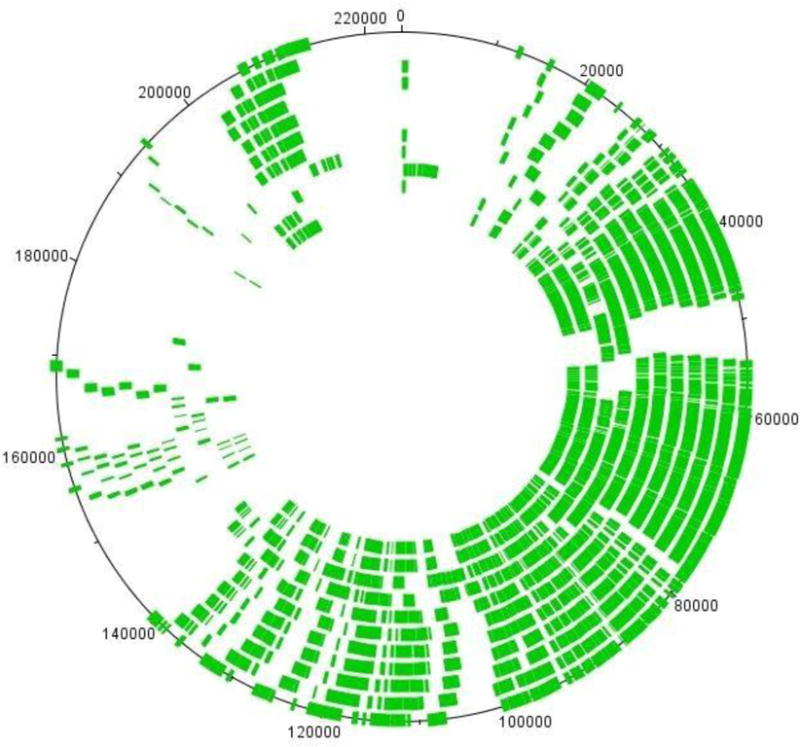
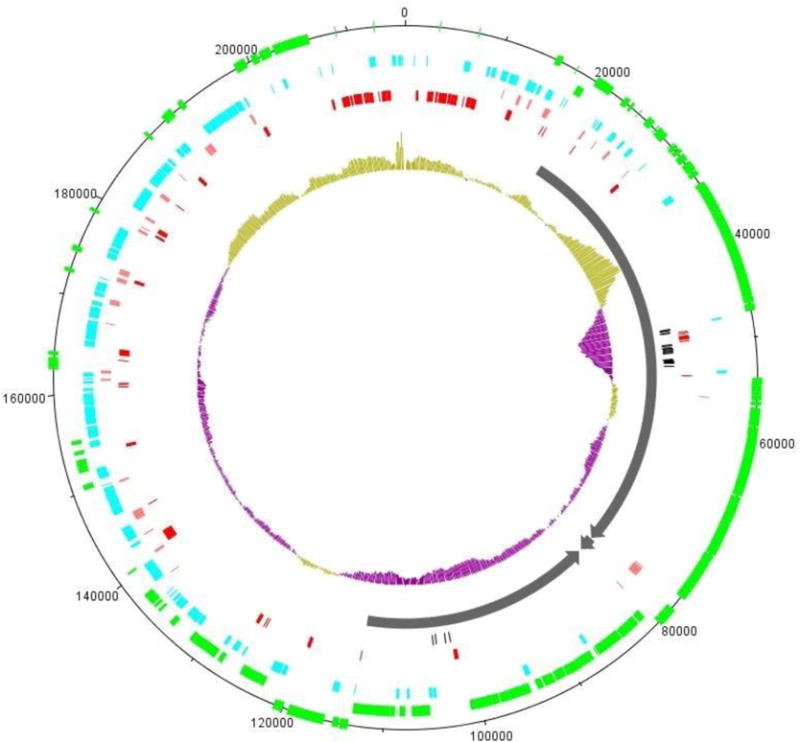
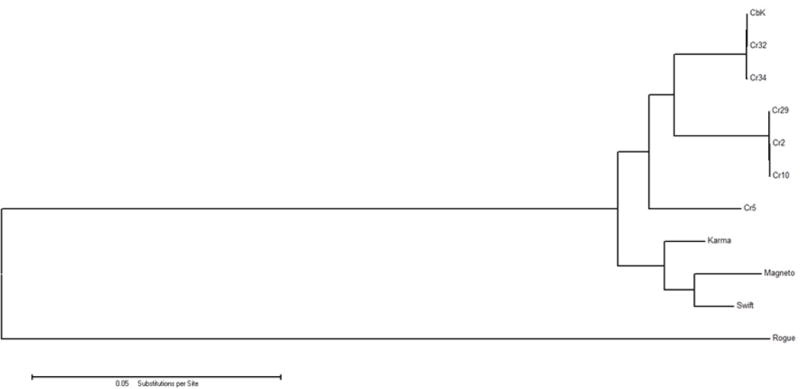
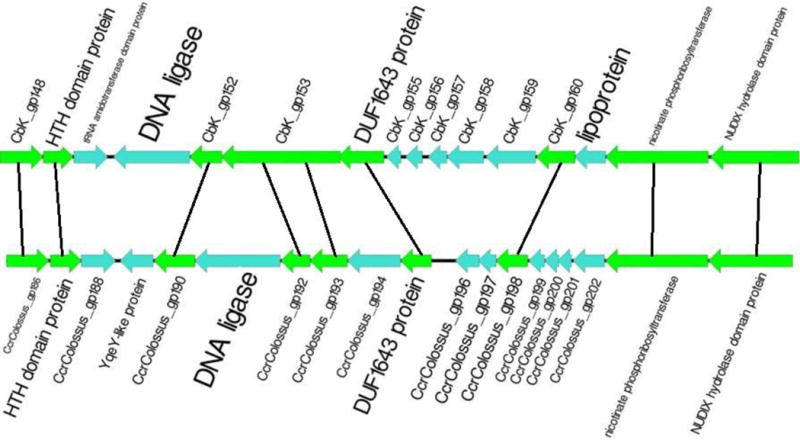
The genomes of the type B3 bacteriophages that infect Caulobacter crescentus are among the largest phage genomes thus far deposited into GenBank with sizes over 200 kb. In this study, we introduce six new bacteriophage genomes which were obtained from phage collected from various water systems in the southeastern United States and from tropical locations across the globe. A comparative analysis of the 12 available genomes revealed a "core genome" which accounts for roughly 1/3 of these bacteriophage genomes and is predominately localized to the head, tail, and lysis gene regions. Despite being isolated from geographically distinct locations, the genomes of these bacteriophages are highly conserved in both genome sequence and gene order. We also identified the insertions, deletions, translocations, and horizontal gene transfer events which are responsible for the genomic diversity of this group of bacteriophages and demonstrated that these changes are not consistent with the idea that modular reassortment of genomes occurs in this group of bacteriophages.
Conflict of interest statement
Figures

Similar articles
-
Analyses of four new Caulobacter Phicbkviruses indicate independent lineages.J Gen Virol. 2019 Feb;100(2):321-331. doi: 10.1099/jgv.0.001218. Epub 2019 Jan 18. J Gen Virol. 2019. PMID: 30657445 Free PMC article.
-
Identification of proteins associated with two diverse Caulobacter phicbkvirus particles.Arch Virol. 2020 Sep;165(9):1995-2002. doi: 10.1007/s00705-020-04707-2. Epub 2020 Jun 25. Arch Virol. 2020. PMID: 32588241 Free PMC article.
-
The Caulobacter crescentus phage phiCbK: genomics of a canonical phage.BMC Genomics. 2012 Oct 10;13:542. doi: 10.1186/1471-2164-13-542. BMC Genomics. 2012. PMID: 23050599 Free PMC article.
-
Bacteriophages and their genomes.Curr Opin Virol. 2011 Oct;1(4):298-303. doi: 10.1016/j.coviro.2011.06.009. Curr Opin Virol. 2011. PMID: 22034588 Free PMC article. Review.
-
Progress on phage genomics of Pseudomonas spp.Yi Chuan. 2020 Aug 20;42(8):752-759. doi: 10.16288/j.yczz.19-272. Yi Chuan. 2020. PMID: 32952111 Review.
Cited by
-
Novel Caulobacter bacteriophages illustrate the diversity of the podovirus genus Rauchvirus.Arch Virol. 2020 Nov;165(11):2549-2554. doi: 10.1007/s00705-020-04791-4. Epub 2020 Sep 1. Arch Virol. 2020. PMID: 32870405 Free PMC article.
-
Analyses of four new Caulobacter Phicbkviruses indicate independent lineages.J Gen Virol. 2019 Feb;100(2):321-331. doi: 10.1099/jgv.0.001218. Epub 2019 Jan 18. J Gen Virol. 2019. PMID: 30657445 Free PMC article.
-
A Novel Phage Infecting the Marine Photoheterotrophic Bacterium Citromicrobium bathyomarinum.Viruses. 2022 Mar 2;14(3):512. doi: 10.3390/v14030512. Viruses. 2022. PMID: 35336919 Free PMC article.
-
Structural and Genomic Diversity of Bacteriophages.Methods Mol Biol. 2024;2738:3-16. doi: 10.1007/978-1-0716-3549-0_1. Methods Mol Biol. 2024. PMID: 37966589 Review.
-
Genomic diversity of bacteriophages infecting Rhodobacter capsulatus and their relatedness to its gene transfer agent RcGTA.PLoS One. 2021 Nov 18;16(11):e0255262. doi: 10.1371/journal.pone.0255262. eCollection 2021. PLoS One. 2021. PMID: 34793465 Free PMC article.
References
-
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi:1471-2164-9-75 [pii] 10.1186/1471-2164- 9-75. - PMC - PubMed
-
- Botstein D. A theory of modular evolution for bacteriophages. Ann N Y Acad Sci. 1980;354:484–490. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
