

MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics
- PMID: 28394336
- PMCID: PMC5409104
- DOI: 10.1038/nmeth.4256
MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics
Abstract
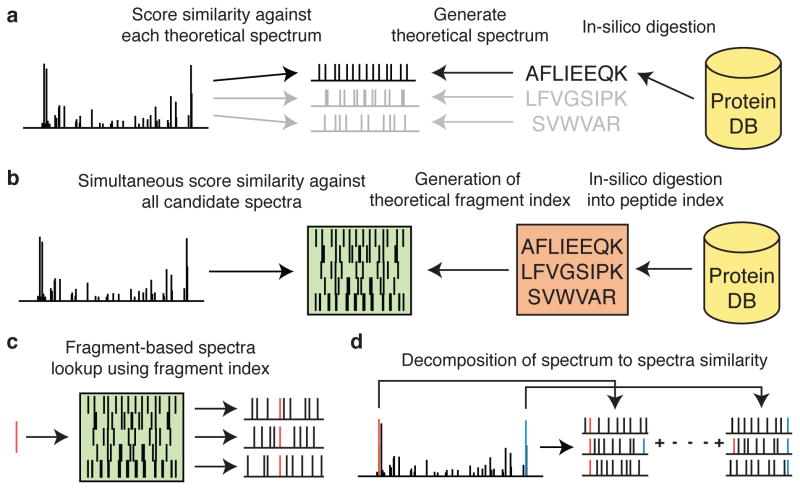
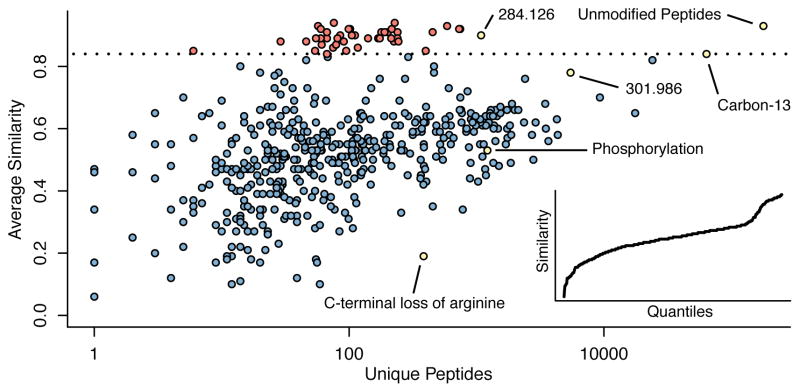
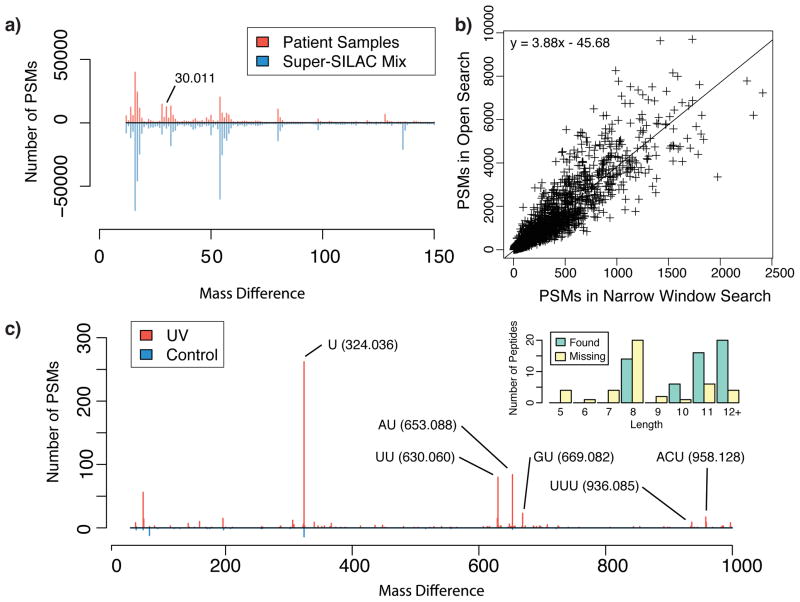
There is a need to better understand and handle the 'dark matter' of proteomics-the vast diversity of post-translational and chemical modifications that are unaccounted in a typical mass spectrometry-based analysis and thus remain unidentified. We present a fragment-ion indexing method, and its implementation in peptide identification tool MSFragger, that enables a more than 100-fold improvement in speed over most existing proteome database search tools. Using several large proteomic data sets, we demonstrate how MSFragger empowers the open database search concept for comprehensive identification of peptides and all their modified forms, uncovering dramatic differences in modification rates across experimental samples and conditions. We further illustrate its utility using protein-RNA cross-linked peptide data and using affinity purification experiments where we observe, on average, a 300% increase in the number of identified spectra for enriched proteins. We also discuss the benefits of open searching for improved false discovery rate estimation in proteomics.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


References
-
- Skinner OS, Kelleher NL. Illuminating the dark matter of shotgun proteomics. Nat Biotech. 2015;33:717–718. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
