

Spatial heterogeneity in medulloblastoma
- PMID: 28394352
- PMCID: PMC5553617
- DOI: 10.1038/ng.3838
Spatial heterogeneity in medulloblastoma
Abstract
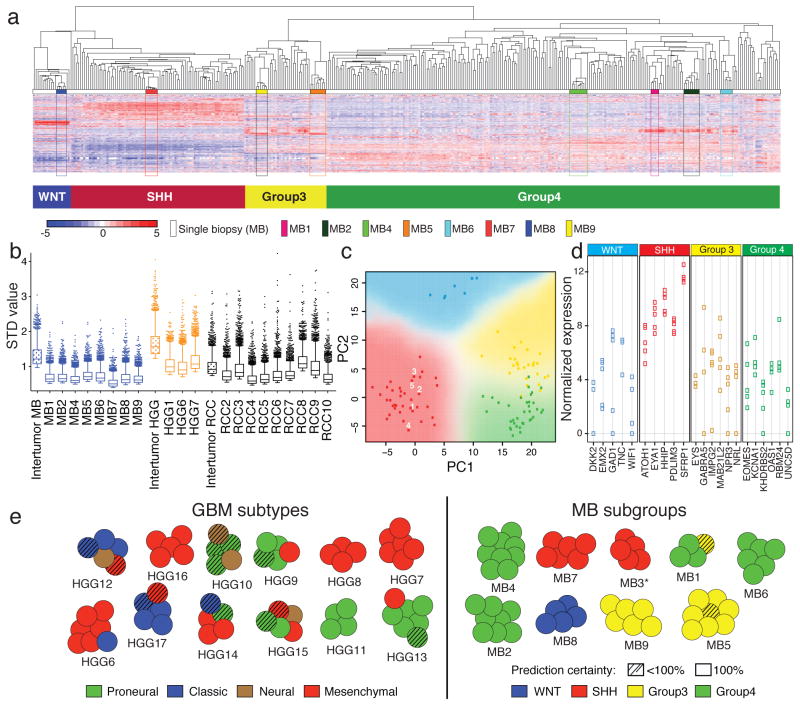
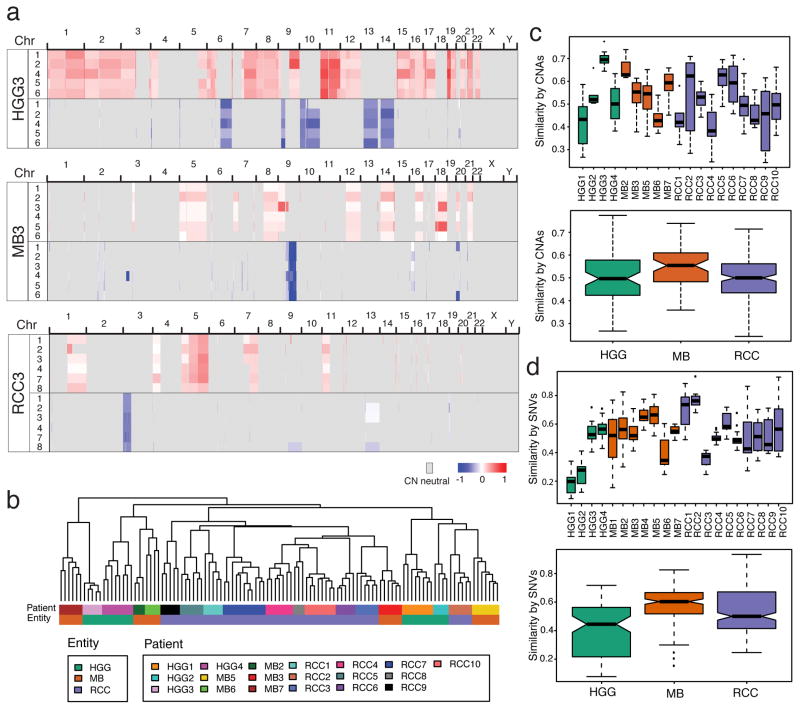
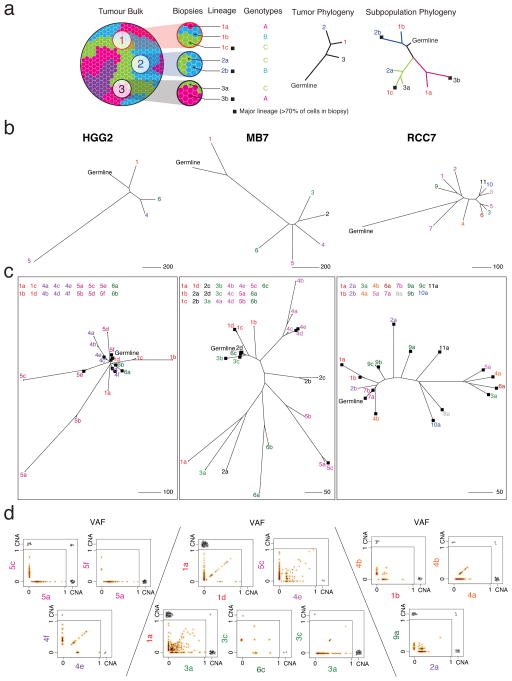
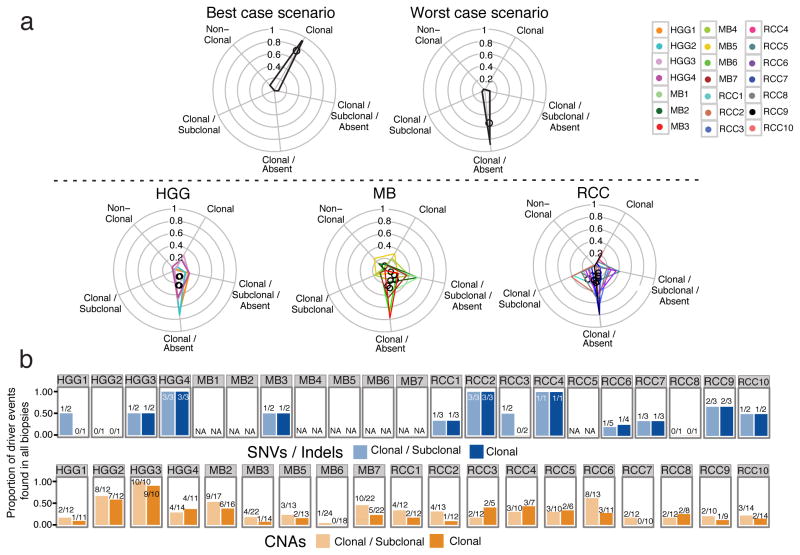
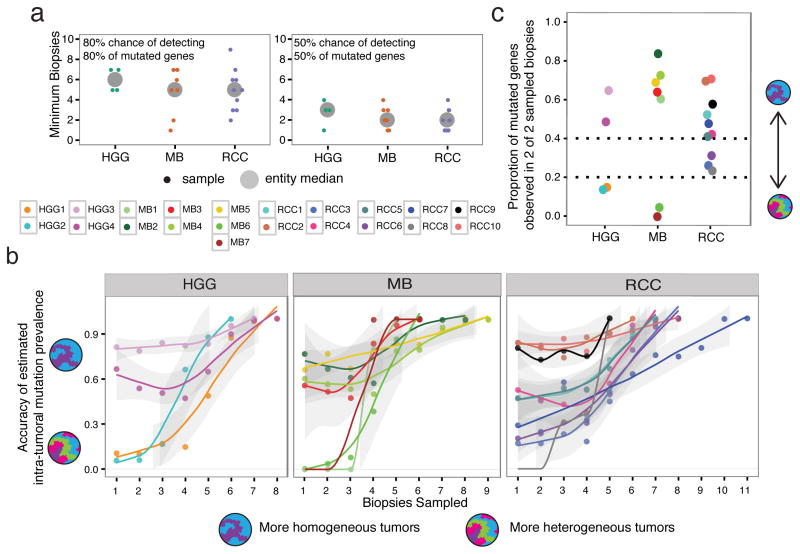
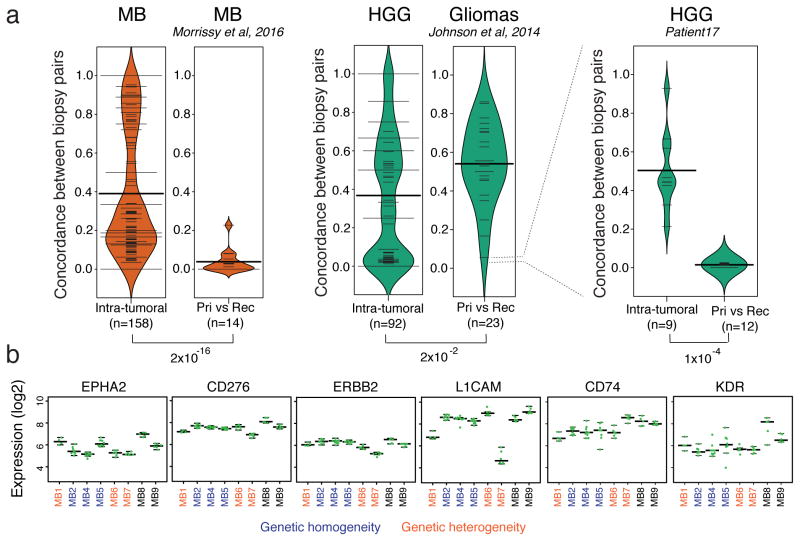
Spatial heterogeneity of transcriptional and genetic markers between physically isolated biopsies of a single tumor poses major barriers to the identification of biomarkers and the development of targeted therapies that will be effective against the entire tumor. We analyzed the spatial heterogeneity of multiregional biopsies from 35 patients, using a combination of transcriptomic and genomic profiles. Medulloblastomas (MBs), but not high-grade gliomas (HGGs), demonstrated spatially homogeneous transcriptomes, which allowed for accurate subgrouping of tumors from a single biopsy. Conversely, somatic mutations that affect genes suitable for targeted therapeutics demonstrated high levels of spatial heterogeneity in MB, malignant glioma, and renal cell carcinoma (RCC). Actionable targets found in a single MB biopsy were seldom clonal across the entire tumor, which brings the efficacy of monotherapies against a single target into question. Clinical trials of targeted therapies for MB should first ensure the spatially ubiquitous nature of the target mutation.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
