

Metabolism in cardiomyopathy: every substrate matters
- PMID: 28395011
- PMCID: PMC5852620
- DOI: 10.1093/cvr/cvx017
Metabolism in cardiomyopathy: every substrate matters
Abstract
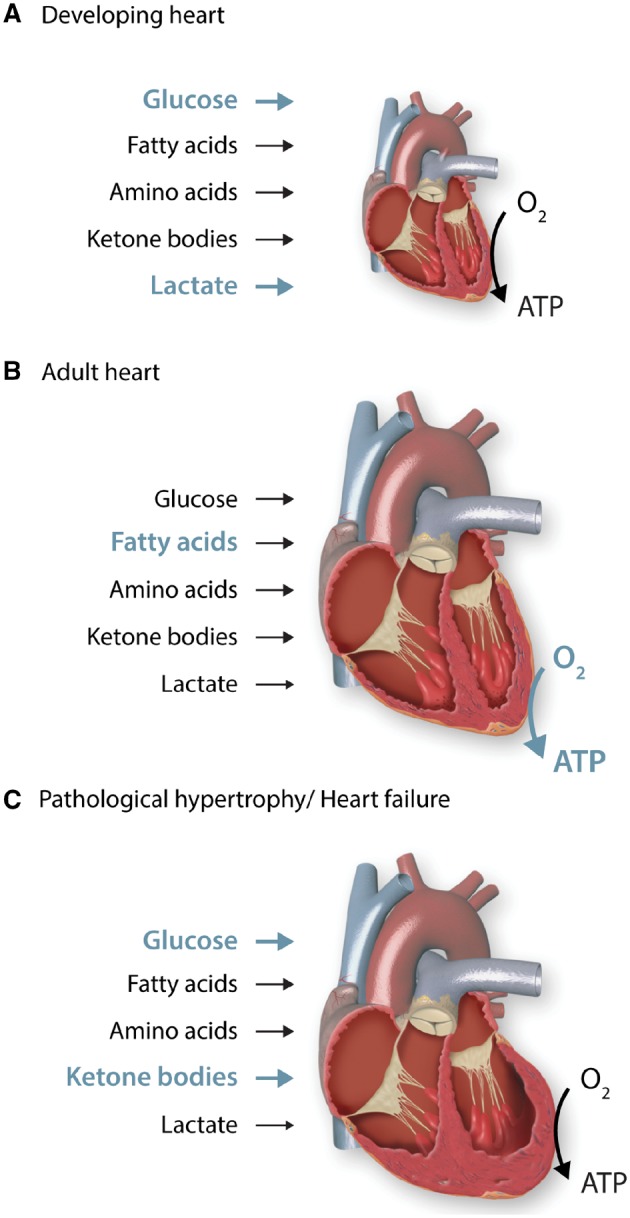
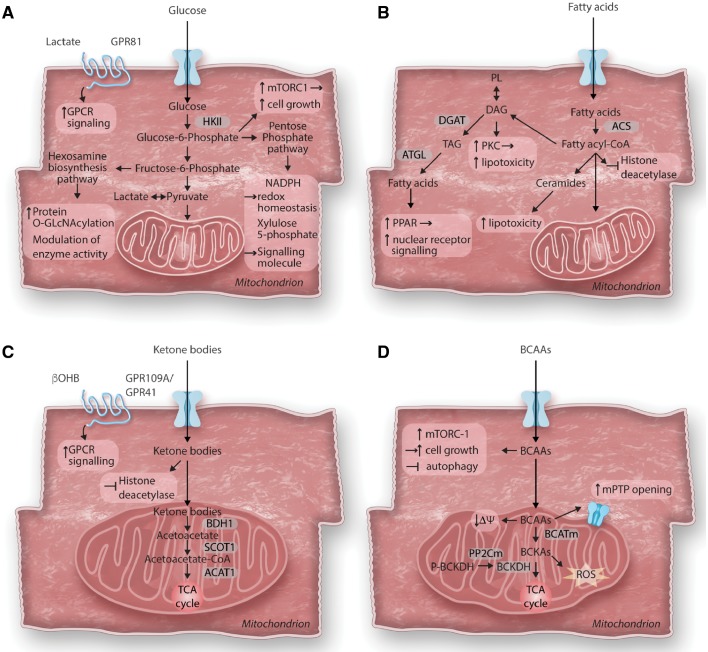
Cardiac metabolism is highly adaptive to changes in fuel availability and the energy demand of the heart. This metabolic flexibility is key for the heart to maintain its output during the development and in response to stress. Alterations in substrate preference have been observed in multiple disease states; a clear understanding of their impact on cardiac function in the long term is critical for the development of metabolic therapies. In addition, the contribution of cellular metabolism to growth, survival, and other signalling pathways through the generation of metabolic intermediates has been increasingly noted, adding another layer of complexity to the impact of metabolism on cardiac function. In a quest to understand the complexity of the cardiac metabolic network, genetic tools have been engaged to manipulate cardiac metabolism in a variety of mouse models. The ability to engineer cardiac metabolism in vivo has provided tremendous insights and brought about conceptual innovations. In this review, we will provide an overview of the cardiac metabolic network and highlight alterations observed during cardiac development and pathological hypertrophy. We will focus on consequences of altered substrate preference on cardiac response to chronic stresses through energy providing and non-energy providing pathways.
Keywords: Cardiac metabolism; Energy metabolism; Metabolic flexibility; Metabolic signalling; Pathological hypertrophy.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author 2017. For permissions, please email: journals.permissions@oup.com.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
