

High-confidence coding and noncoding transcriptome maps
- PMID: 28396519
- PMCID: PMC5453319
- DOI: 10.1101/gr.214288.116
High-confidence coding and noncoding transcriptome maps
Abstract
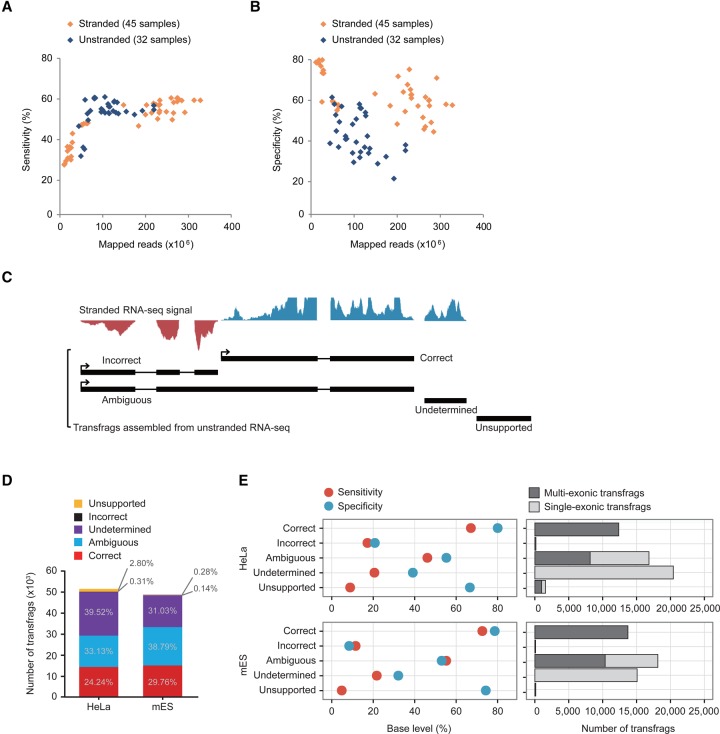
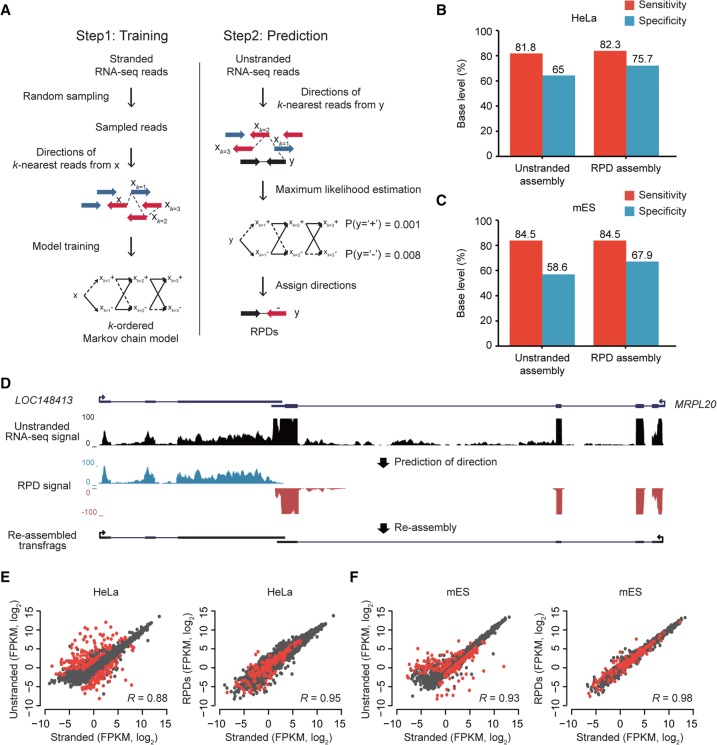
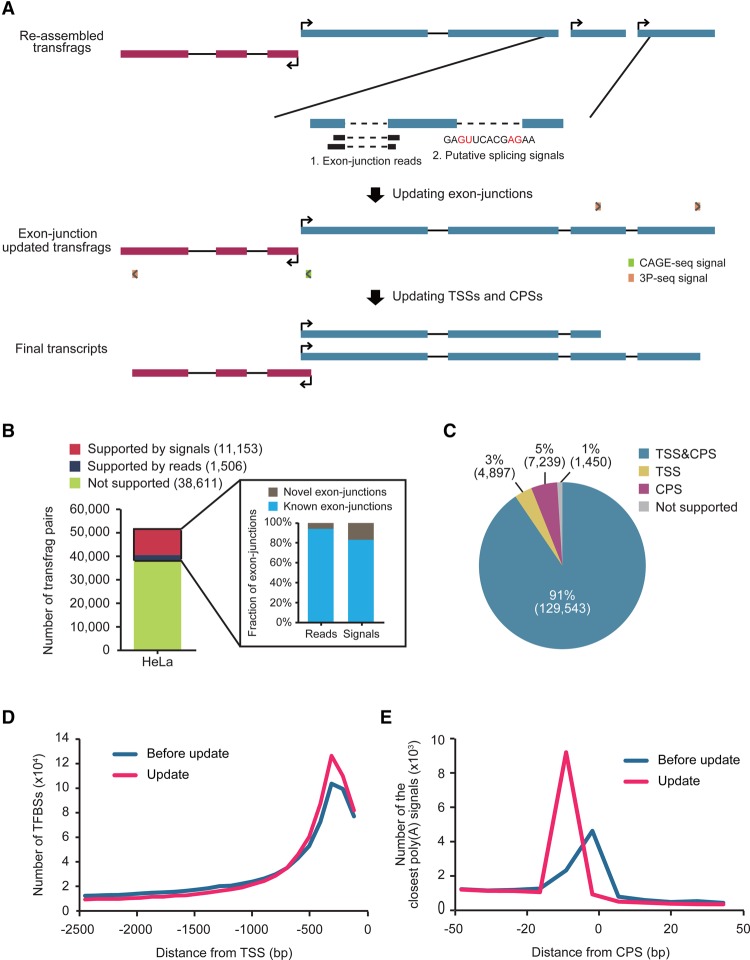
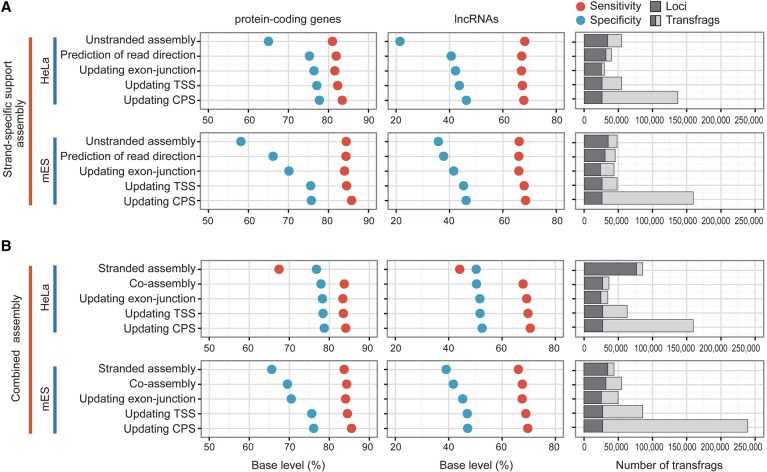
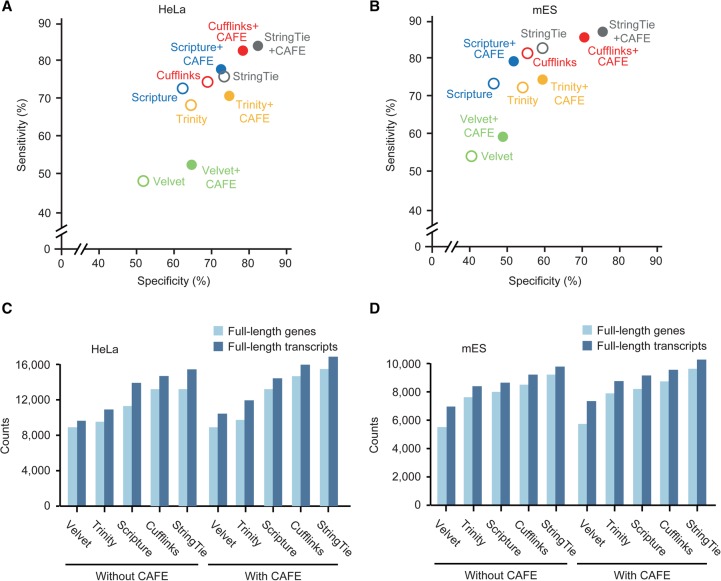
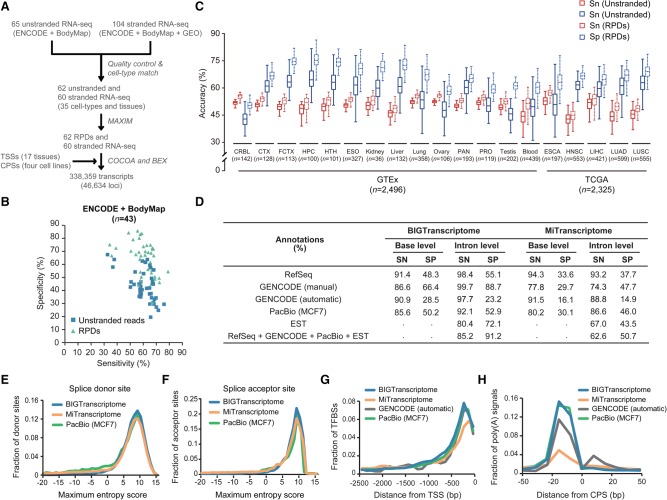
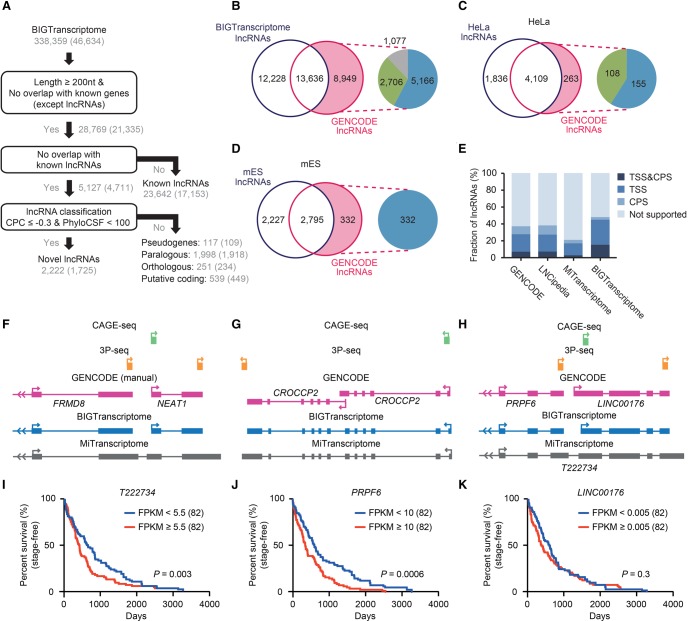
The advent of high-throughput RNA sequencing (RNA-seq) has led to the discovery of unprecedentedly immense transcriptomes encoded by eukaryotic genomes. However, the transcriptome maps are still incomplete partly because they were mostly reconstructed based on RNA-seq reads that lack their orientations (known as unstranded reads) and certain boundary information. Methods to expand the usability of unstranded RNA-seq data by predetermining the orientation of the reads and precisely determining the boundaries of assembled transcripts could significantly benefit the quality of the resulting transcriptome maps. Here, we present a high-performing transcriptome assembly pipeline, called CAFE, that significantly improves the original assemblies, respectively assembled with stranded and/or unstranded RNA-seq data, by orienting unstranded reads using the maximum likelihood estimation and by integrating information about transcription start sites and cleavage and polyadenylation sites. Applying large-scale transcriptomic data comprising 230 billion RNA-seq reads from the ENCODE, Human BodyMap 2.0, The Cancer Genome Atlas, and GTEx projects, CAFE enabled us to predict the directions of about 220 billion unstranded reads, which led to the construction of more accurate transcriptome maps, comparable to the manually curated map, and a comprehensive lncRNA catalog that includes thousands of novel lncRNAs. Our pipeline should not only help to build comprehensive, precise transcriptome maps from complex genomes but also to expand the universe of noncoding genomes.
© 2017 You et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases