

Trimming the Vascular Tree in Tumors: Metabolic and Immune Adaptations
- PMID: 28396525
- PMCID: PMC8335596
- DOI: 10.1101/sqb.2016.81.030940
Trimming the Vascular Tree in Tumors: Metabolic and Immune Adaptations
Abstract
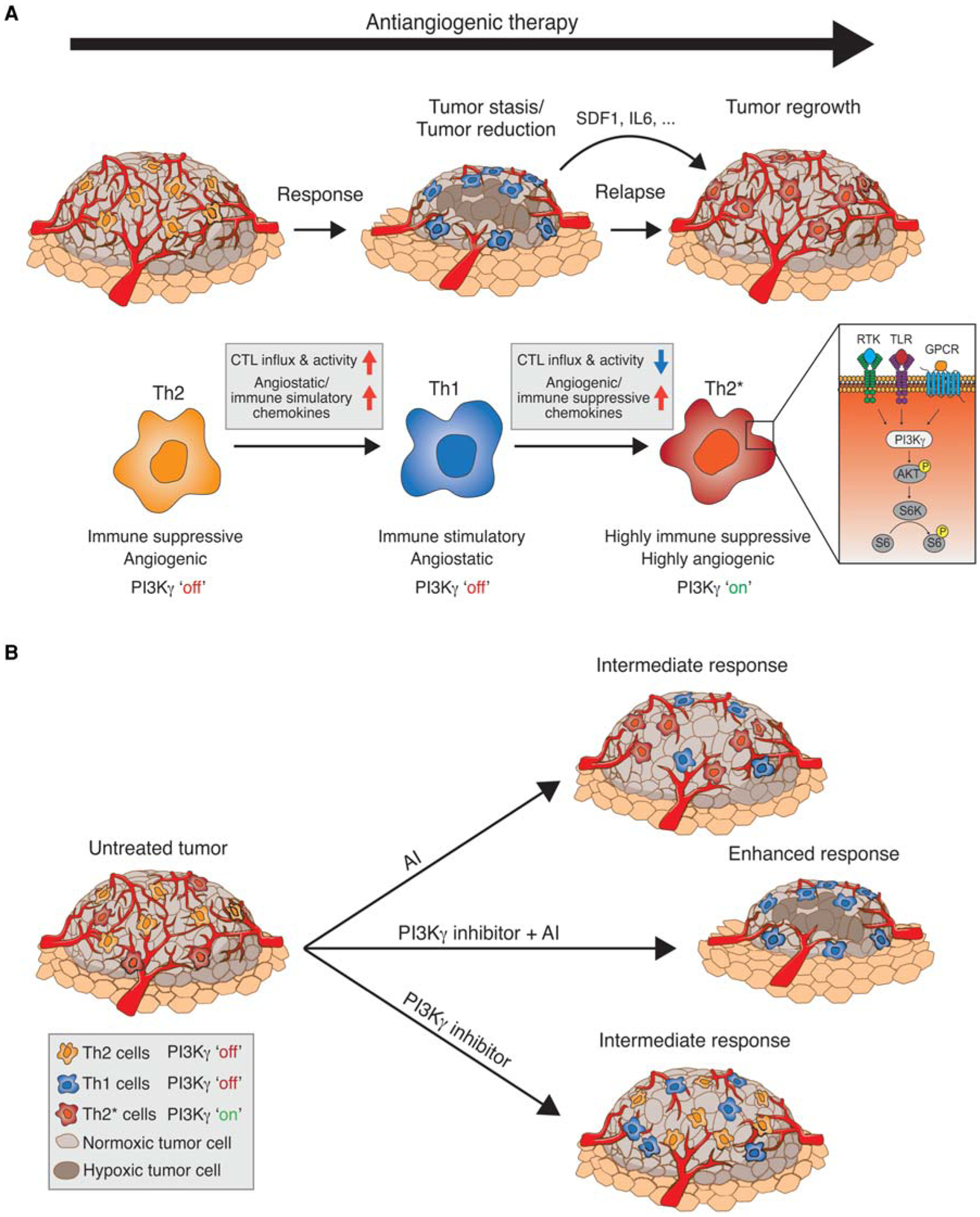
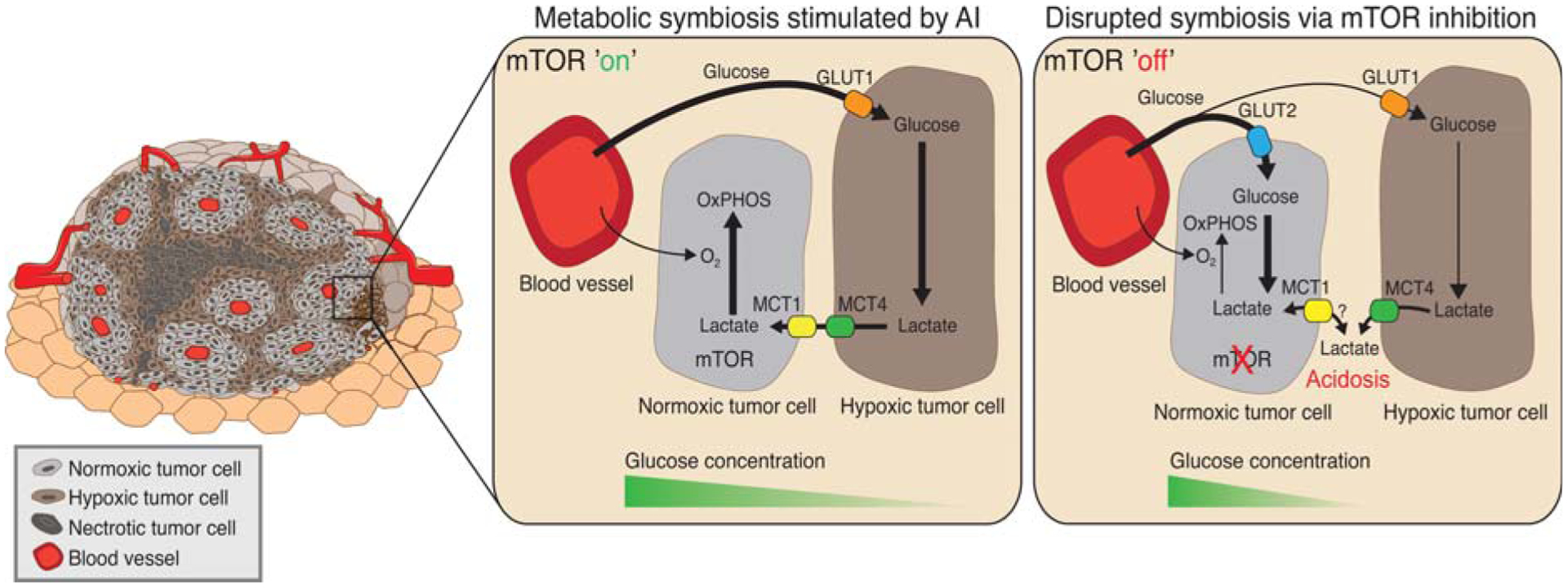
Angiogenesis, the formation of new blood vessels, has become a well-established hallmark of cancer. Its functional importance for the manifestation and progression of tumors has been further validated by the beneficial therapeutic effects of angiogenesis inhibitors, most notably ones targeting the vascular endothelial growth factor (VEGF) signaling pathways. However, with the transient and short-lived nature of the patient response, it has become evident that tumors have the ability to adapt to the pressures of vascular growth restriction. Several escape mechanisms have been described that adapt tumors to therapy-induced low-oxygen tension by either reinstating tumor growth by vascular rebound or by altering tumor behavior without the necessity to reinitiate revascularization. We review here two bypass mechanisms that either instigate angiogenic and immune-suppressive polarization of intratumoral innate immune cells to facilitate VEGF-independent angiogenesis or enable metabolic adaptation and reprogramming of endothelial cells and tumor cells to adapt to low-oxygen tension.
© 2016 Allen et al; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Escape mechanisms from antiangiogenic therapy: an immune cell's perspective.Adv Exp Med Biol. 2014;772:83-99. doi: 10.1007/978-1-4614-5915-6_4. Adv Exp Med Biol. 2014. PMID: 24272355 Review.
-
Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis.Pharmacol Ther. 2018 Nov;191:92-122. doi: 10.1016/j.pharmthera.2018.06.003. Epub 2018 Jun 29. Pharmacol Ther. 2018. PMID: 29909237 Review.
-
Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor.Oncologist. 2015 Jun;20(6):660-73. doi: 10.1634/theoncologist.2014-0465. Epub 2015 May 22. Oncologist. 2015. PMID: 26001391 Free PMC article. Review.
-
Vascular endothelial cell growth factor (VEGF), an emerging target for cancer chemotherapy.Curr Med Chem Anticancer Agents. 2003 Mar;3(2):95-117. doi: 10.2174/1568011033353452. Curr Med Chem Anticancer Agents. 2003. PMID: 12678905 Review.
-
VEGF signaling in cancer treatment.Curr Pharm Des. 2014;20(17):2834-42. doi: 10.2174/13816128113199990590. Curr Pharm Des. 2014. PMID: 23944367 Review.
Cited by
-
βIV-spectrin as a stalk cell-intrinsic regulator of VEGF signaling.Nat Commun. 2022 Mar 14;13(1):1326. doi: 10.1038/s41467-022-28933-1. Nat Commun. 2022. PMID: 35288568 Free PMC article.
-
Targeting Angiogenesis With Peptide Vaccines.Front Immunol. 2019 Aug 8;10:1924. doi: 10.3389/fimmu.2019.01924. eCollection 2019. Front Immunol. 2019. PMID: 31440262 Free PMC article. Review.
-
Editorial: Targeting Angiogenesis to Treat Autoimmune Diseases and Cancer.Front Immunol. 2020 May 21;11:1005. doi: 10.3389/fimmu.2020.01005. eCollection 2020. Front Immunol. 2020. PMID: 32528478 Free PMC article. No abstract available.
References
-
- Adams RH, Alitalo K. 2007. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478. - PubMed
-
- Baluk P, Hashizume H, McDonald DM. 2005. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15: 102–111. - PubMed
-
- Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, Ancukiewicz M, Polaskova P, Pinho MC, Jennings D, et al. 2013. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci 110: 19059–19064. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources