

Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis
- PMID: 28398466
- PMCID: PMC5458344
- DOI: 10.1093/hmg/ddx116
Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis
Abstract
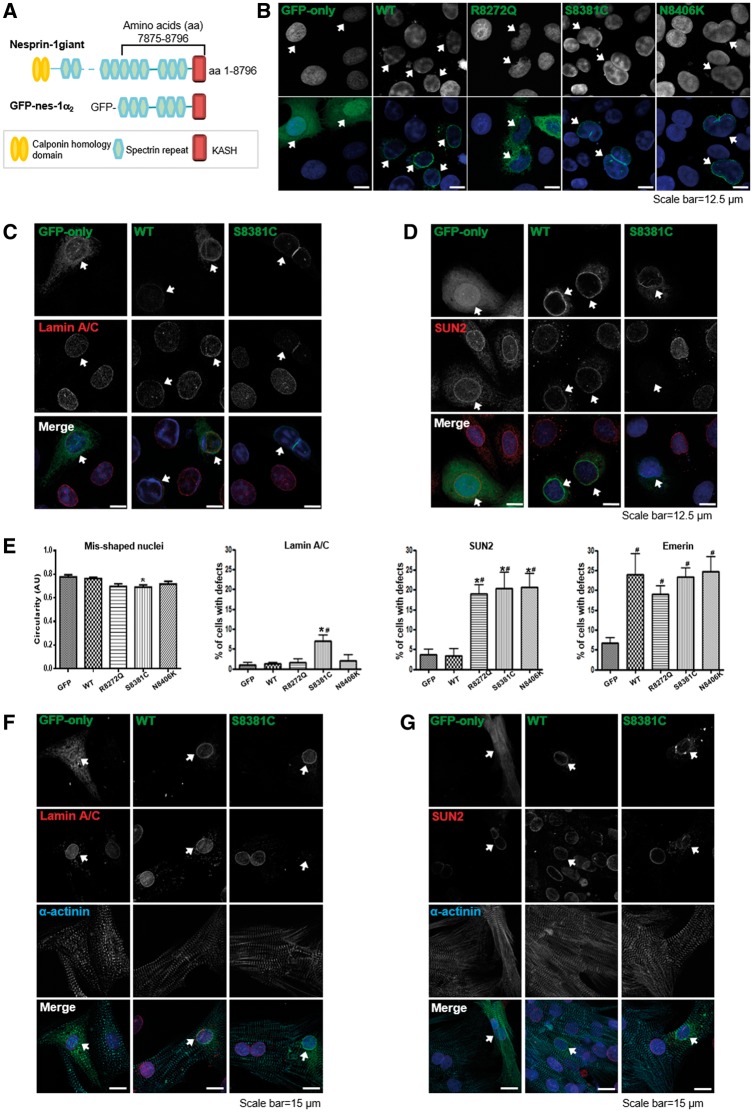
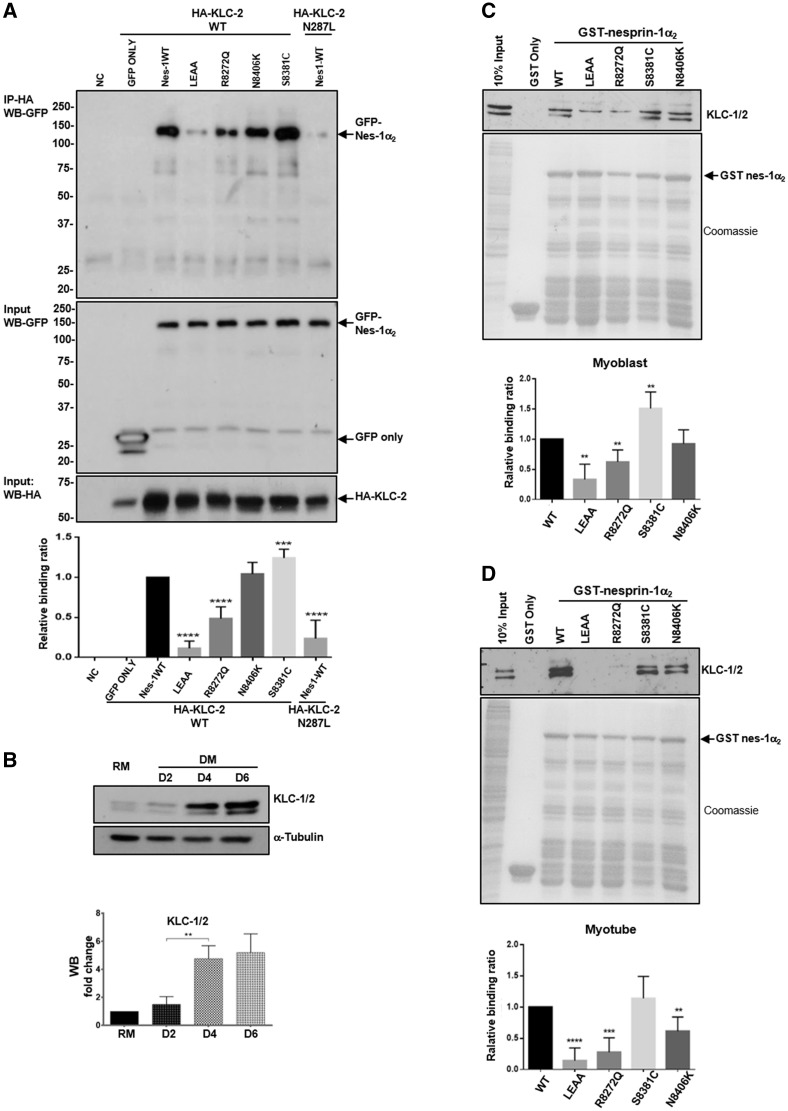
Nesprins-1 and -2 are highly expressed in skeletal and cardiac muscle and together with SUN (Sad1p/UNC84)-domain containing proteins and lamin A/C form the LInker of Nucleoskeleton-and-Cytoskeleton (LINC) bridging complex at the nuclear envelope (NE). Mutations in nesprin-1/2 have previously been found in patients with autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD) as well as dilated cardiomyopathy (DCM). In this study, three novel rare variants (R8272Q, S8381C and N8406K) in the C-terminus of the SYNE1 gene (nesprin-1) were identified in seven DCM patients by mutation screening. Expression of these mutants caused nuclear morphology defects and reduced lamin A/C and SUN2 staining at the NE. GST pull-down indicated that nesprin-1/lamin/SUN interactions were disrupted. Nesprin-1 mutations were also associated with augmented activation of the ERK pathway in vitro and in hearts in vivo. During C2C12 muscle cell differentiation, nesprin-1 levels are increased concomitantly with kinesin light chain (KLC-1/2) and immunoprecipitation and GST pull-down showed that these proteins interacted via a recently identified LEWD domain in the C-terminus of nesprin-1. Expression of nesprin-1 mutants in C2C12 cells caused defects in myoblast differentiation and fusion associated with dysregulation of myogenic transcription factors and disruption of the nesprin-1 and KLC-1/2 interaction at the outer nuclear membrane. Expression of nesprin-1α2 WT and mutants in zebrafish embryos caused heart developmental defects that varied in severity. These findings support a role for nesprin-1 in myogenesis and muscle disease, and uncover a novel mechanism whereby disruption of the LINC complex may contribute to the pathogenesis of DCM.
© The Author 2017. Published by Oxford University Press.
Figures









Similar articles
-
Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity.Hum Mol Genet. 2007 Dec 1;16(23):2816-33. doi: 10.1093/hmg/ddm238. Epub 2007 Aug 29. Hum Mol Genet. 2007. PMID: 17761684
-
Nesprin-1/2: roles in nuclear envelope organisation, myogenesis and muscle disease.Biochem Soc Trans. 2018 Apr 17;46(2):311-320. doi: 10.1042/BST20170149. Epub 2018 Feb 27. Biochem Soc Trans. 2018. PMID: 29487227 Review.
-
Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice.Hum Mol Genet. 2009 Feb 15;18(4):607-20. doi: 10.1093/hmg/ddn386. Epub 2008 Nov 13. Hum Mol Genet. 2009. PMID: 19008300 Free PMC article.
-
LINC complex alterations in DMD and EDMD/CMT fibroblasts.Eur J Cell Biol. 2012 Aug;91(8):614-28. doi: 10.1016/j.ejcb.2012.03.003. Epub 2012 May 1. Eur J Cell Biol. 2012. PMID: 22555292 Free PMC article.
-
Nesprin proteins: bridging nuclear envelope dynamics to muscular dysfunction.Cell Commun Signal. 2024 Apr 2;22(1):208. doi: 10.1186/s12964-024-01593-y. Cell Commun Signal. 2024. PMID: 38566066 Free PMC article. Review.
Cited by
-
Cross-ancestry genome-wide analysis of atrial fibrillation unveils disease biology and enables cardioembolic risk prediction.Nat Genet. 2023 Feb;55(2):187-197. doi: 10.1038/s41588-022-01284-9. Epub 2023 Jan 19. Nat Genet. 2023. PMID: 36653681 Free PMC article.
-
Ablation of SUN2-containing LINC complexes drives cardiac hypertrophy without interstitial fibrosis.Mol Biol Cell. 2019 Jul 1;30(14):1664-1675. doi: 10.1091/mbc.E18-07-0438. Epub 2019 May 15. Mol Biol Cell. 2019. PMID: 31091167 Free PMC article.
-
Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation.Int J Mol Sci. 2021 Jul 6;22(14):7281. doi: 10.3390/ijms22147281. Int J Mol Sci. 2021. PMID: 34298904 Free PMC article. Review.
-
Nuclear SUN1 stabilizes endothelial cell junctions via microtubules to regulate blood vessel formation.Elife. 2023 Mar 29;12:e83652. doi: 10.7554/eLife.83652. Elife. 2023. PMID: 36989130 Free PMC article.
-
Semantic segmentation of HeLa cells: An objective comparison between one traditional algorithm and four deep-learning architectures.PLoS One. 2020 Oct 2;15(10):e0230605. doi: 10.1371/journal.pone.0230605. eCollection 2020. PLoS One. 2020. PMID: 33006963 Free PMC article.
References
-
- Towbin J.A., Bowles N.E. (2002) The failing heart. Nature, 415, 227–233. - PubMed
-
- Hershberger R.E., Hedges D.J., Morales A. (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol., 10, 531–547. - PubMed
-
- Capell B.C., Collins F.S. (2006) Human laminopathies: nuclei gone genetically awry. Nat. Rev. Genet., 7, 940–952. - PubMed
-
- Bonne G., Di Barletta M.R., Varnous S., Becane H.M., Hammouda E.H., Merlini L., Muntoni F., Greenberg C.R., Gary F., Urtizberea J.A., et al. (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet., 21, 285–288. - PubMed
-
- Bione S., Maestrini E., Rivella S., Mancini M., Regis S., Romeo G., Toniolo D. (1994) Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet., 8, 323–327. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
