

Multi-tissue DNA methylation age predictor in mouse
- PMID: 28399939
- PMCID: PMC5389178
- DOI: 10.1186/s13059-017-1203-5
Multi-tissue DNA methylation age predictor in mouse
Abstract
Background: DNA methylation changes at a discrete set of sites in the human genome are predictive of chronological and biological age. However, it is not known whether these changes are causative or a consequence of an underlying ageing process. It has also not been shown whether this epigenetic clock is unique to humans or conserved in the more experimentally tractable mouse.
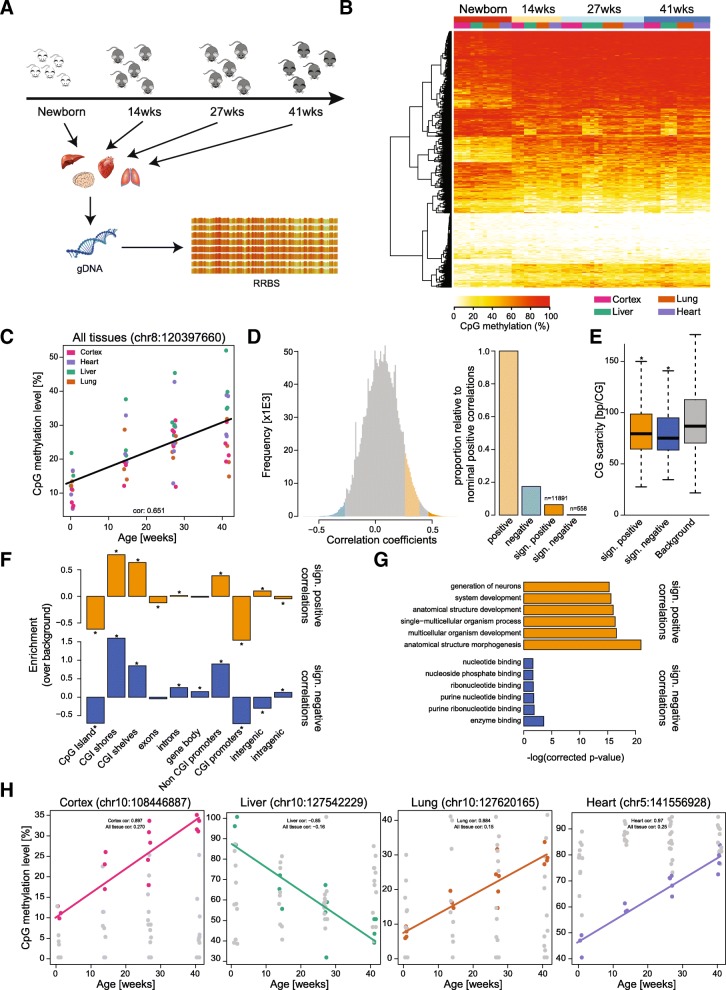
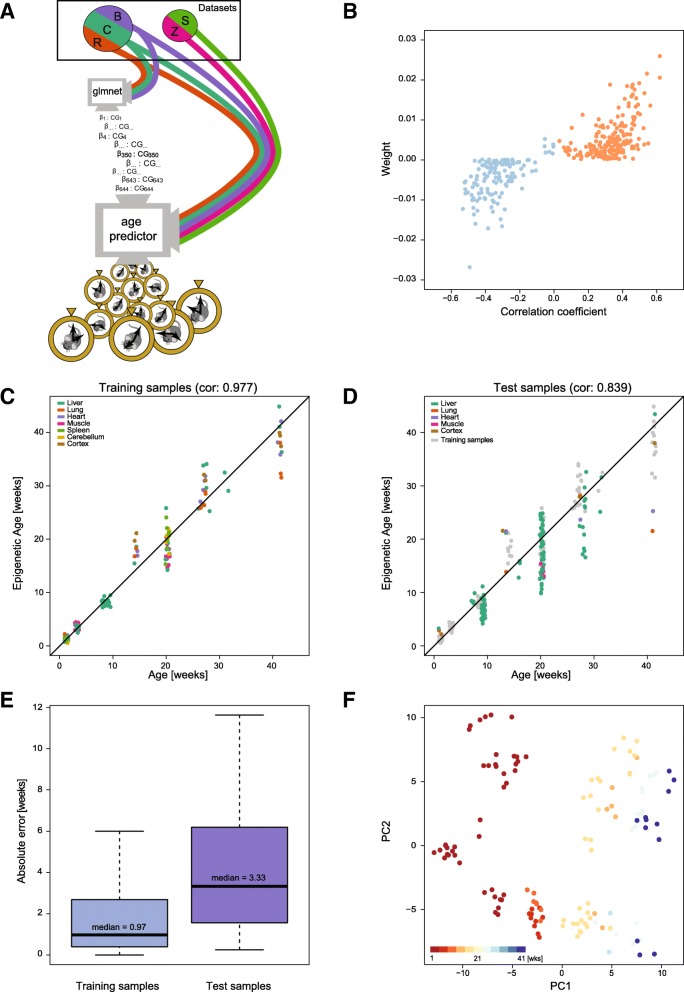
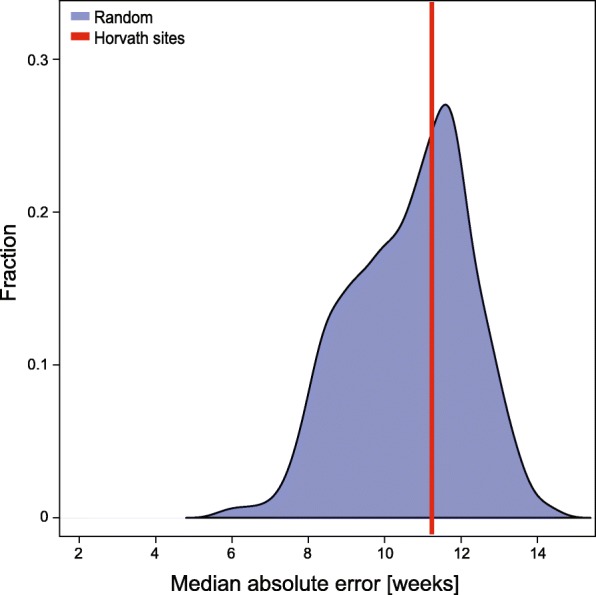
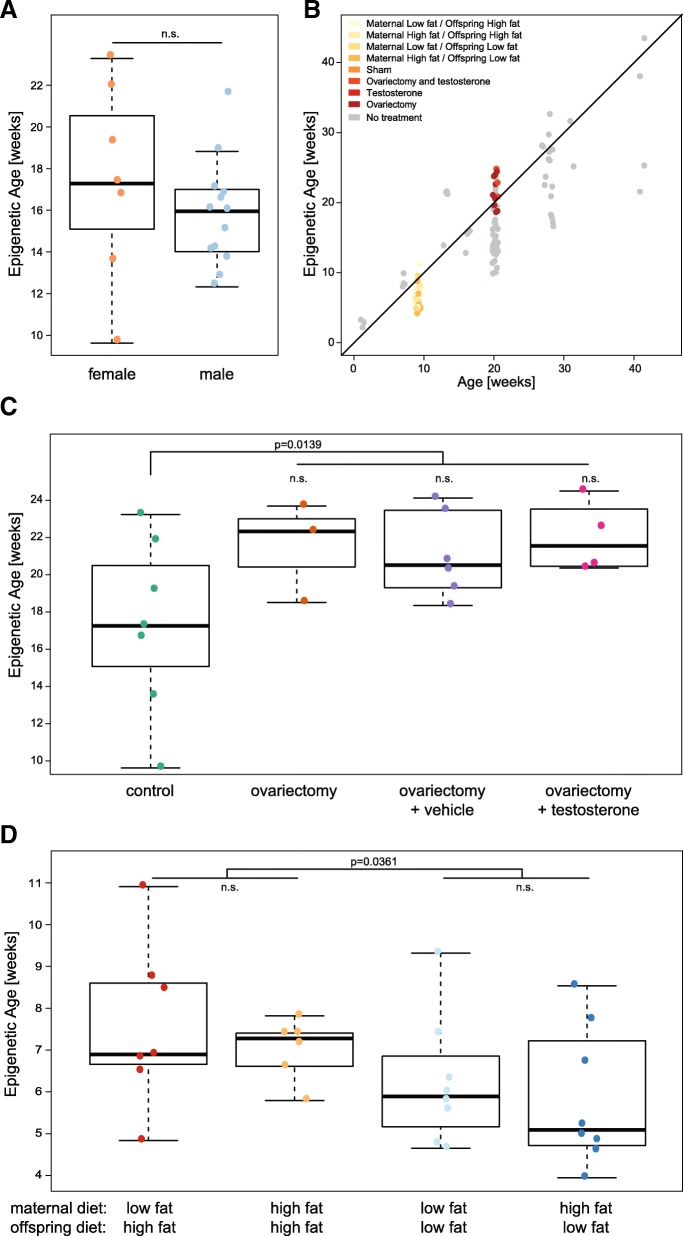
Results: We have generated a comprehensive set of genome-scale base-resolution methylation maps from multiple mouse tissues spanning a wide range of ages. Many CpG sites show significant tissue-independent correlations with age which allowed us to develop a multi-tissue predictor of age in the mouse. Our model, which estimates age based on DNA methylation at 329 unique CpG sites, has a median absolute error of 3.33 weeks and has similar properties to the recently described human epigenetic clock. Using publicly available datasets, we find that the mouse clock is accurate enough to measure effects on biological age, including in the context of interventions. While females and males show no significant differences in predicted DNA methylation age, ovariectomy results in significant age acceleration in females. Furthermore, we identify significant differences in age-acceleration dependent on the lipid content of the diet.
Conclusions: Here we identify and characterise an epigenetic predictor of age in mice, the mouse epigenetic clock. This clock will be instrumental for understanding the biology of ageing and will allow modulation of its ticking rate and resetting the clock in vivo to study the impact on biological age.
Keywords: Ageing/aging; Biological age; Chronological age; DNA methylation; Epigenetic clock; Epigenetics; High fat diet; Model; Ovariectomy; Prediction.
Figures
References
Publication types
MeSH terms
Grants and funding
- BBS/E/B/000C0407/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/B/000C0409/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 095645/Z/11/Z/WT_/Wellcome Trust/United Kingdom
- BB/K010867/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
