

Cancer Manipulation of Host Physiology: Lessons from Pancreatic Cancer
- PMID: 28400243
- PMCID: PMC5480288
- DOI: 10.1016/j.molmed.2017.03.003
Cancer Manipulation of Host Physiology: Lessons from Pancreatic Cancer
Abstract
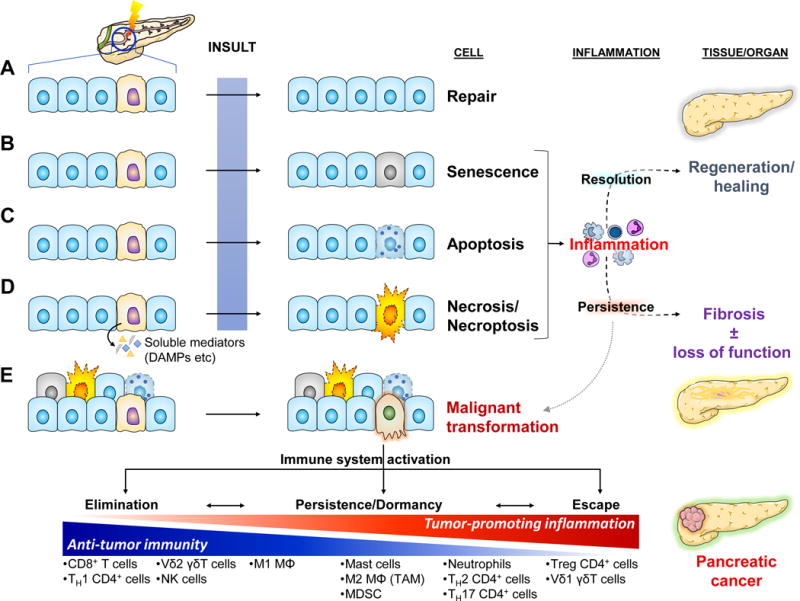
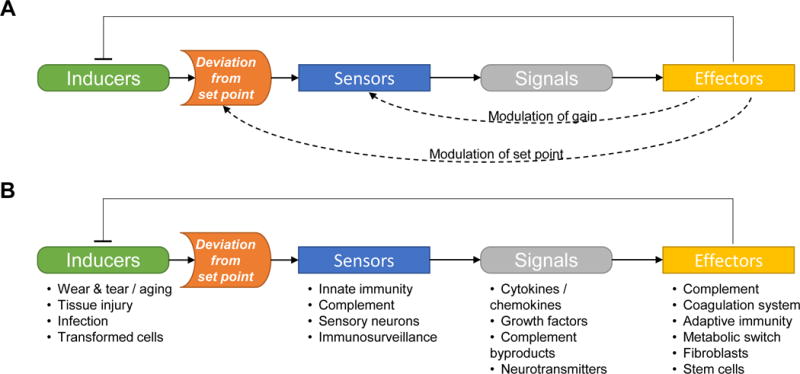
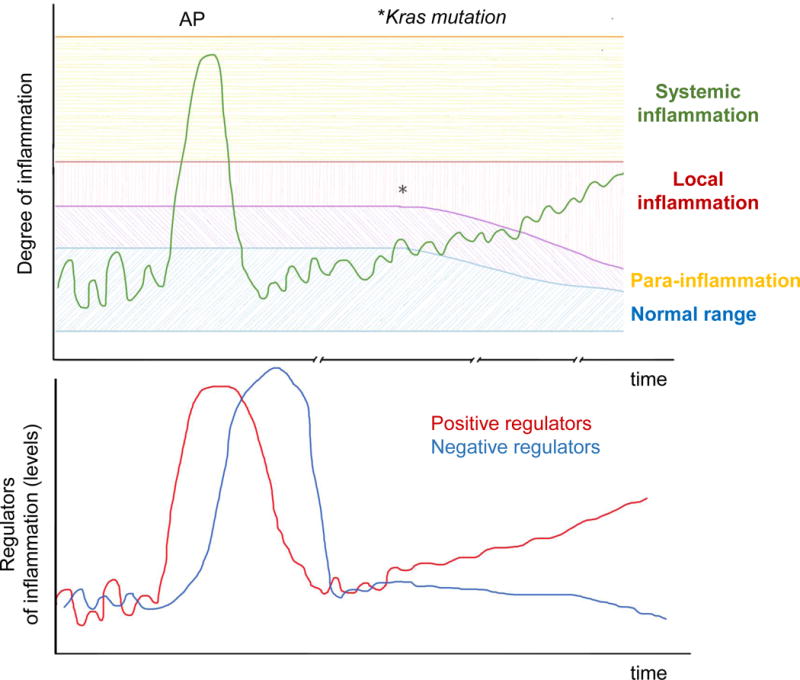
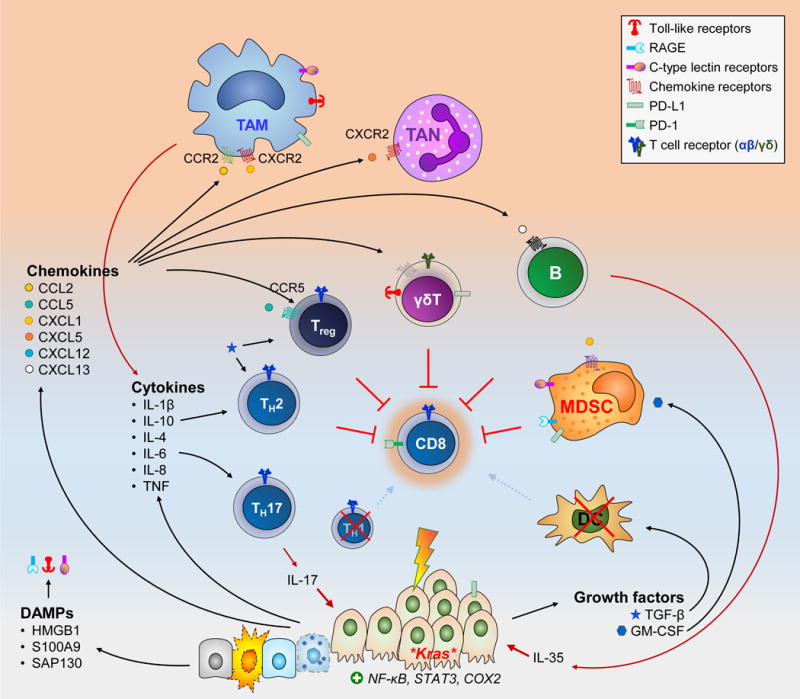
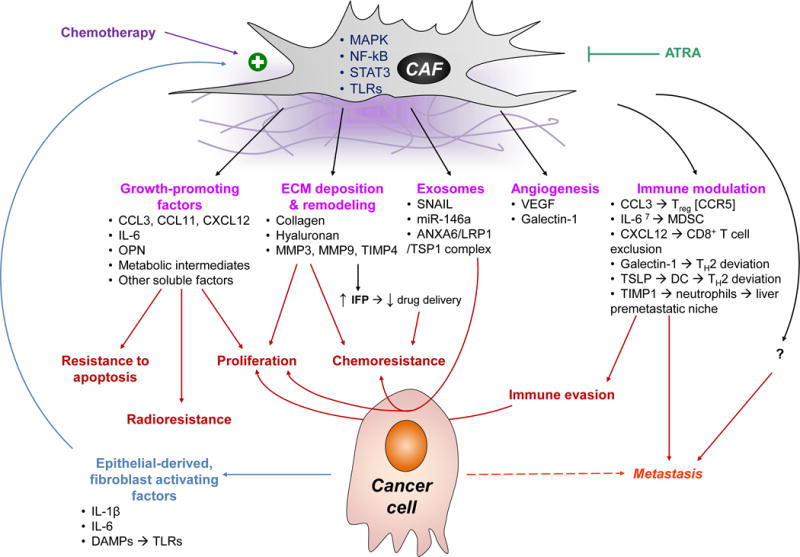
Homeostasis is a fundamental property of living organisms enabling the human body to withstand internal and external insults. In several chronic diseases, and especially in cancer, many homeostatic mechanisms are deranged. Pancreatic cancer in particular is notorious for its ability to invoke an intense fibroinflammatory stromal reaction facilitating its progression and resistance to treatment. In the past decade, several seminal discoveries have elucidated previously unrecognized modes of commandeering the host's defense systems. Here we review novel discoveries in pancreatic cancer immunobiology and attempt to integrate the notion of deranged homeostasis in the pathogenesis of this disease. We also highlight areas of controversy and obstacles that need to be overcome, hoping to further our mechanistic insight into this malignancy.
Keywords: cancer-associated fibroblasts; immunity; inflammation; pancreatic ductal adenocarcinoma; stroma.
Published by Elsevier Ltd.
Conflict of interest statement
None declared.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
