

Deletion of TRPC6 Attenuates NMDA Receptor-Mediated Ca2+ Entry and Ca2+-Induced Neurotoxicity Following Cerebral Ischemia and Oxygen-Glucose Deprivation
- PMID: 28400714
- PMCID: PMC5368256
- DOI: 10.3389/fnins.2017.00138
Deletion of TRPC6 Attenuates NMDA Receptor-Mediated Ca2+ Entry and Ca2+-Induced Neurotoxicity Following Cerebral Ischemia and Oxygen-Glucose Deprivation
Abstract
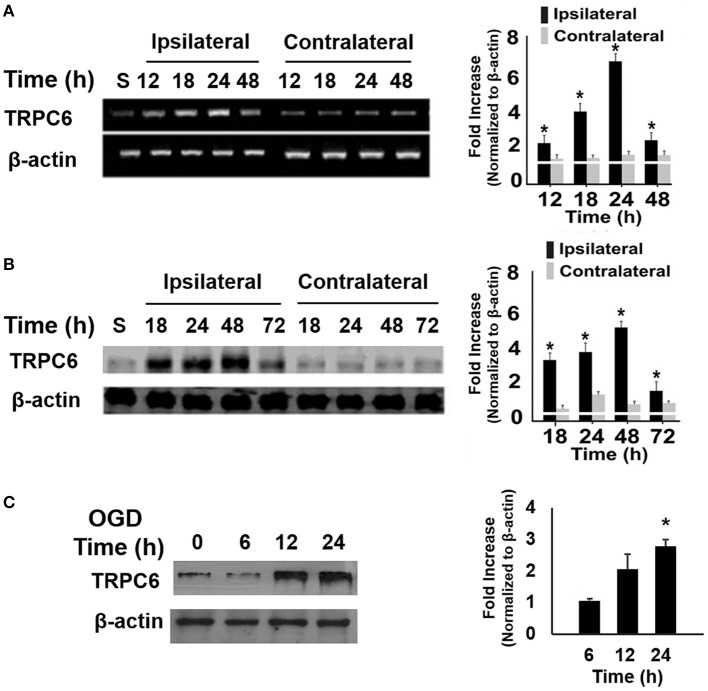
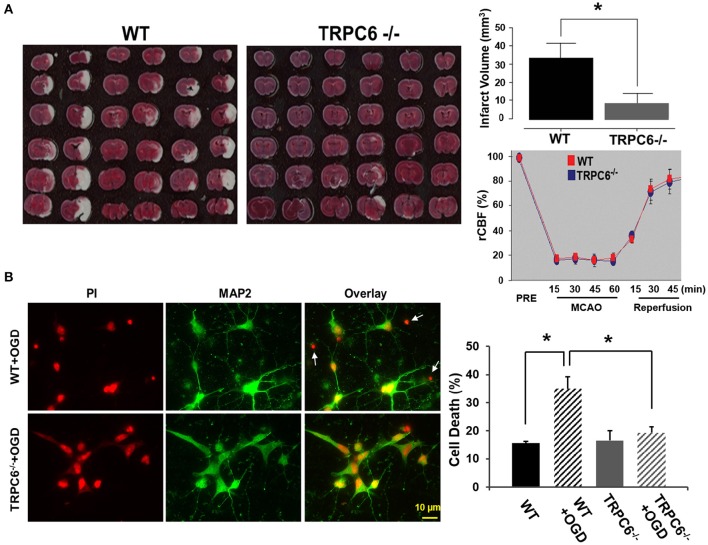
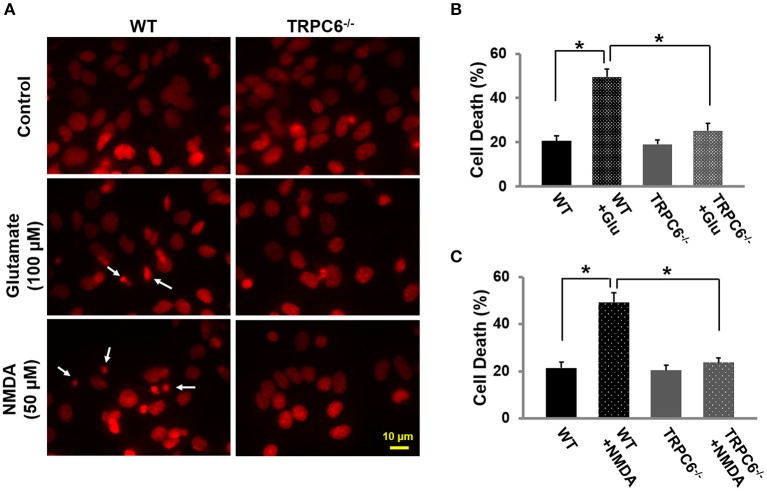
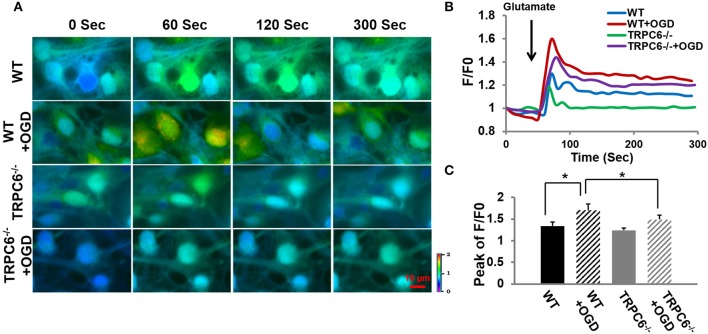
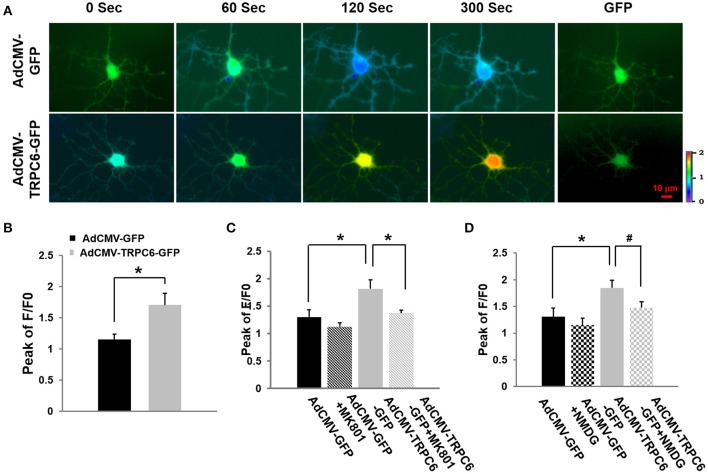
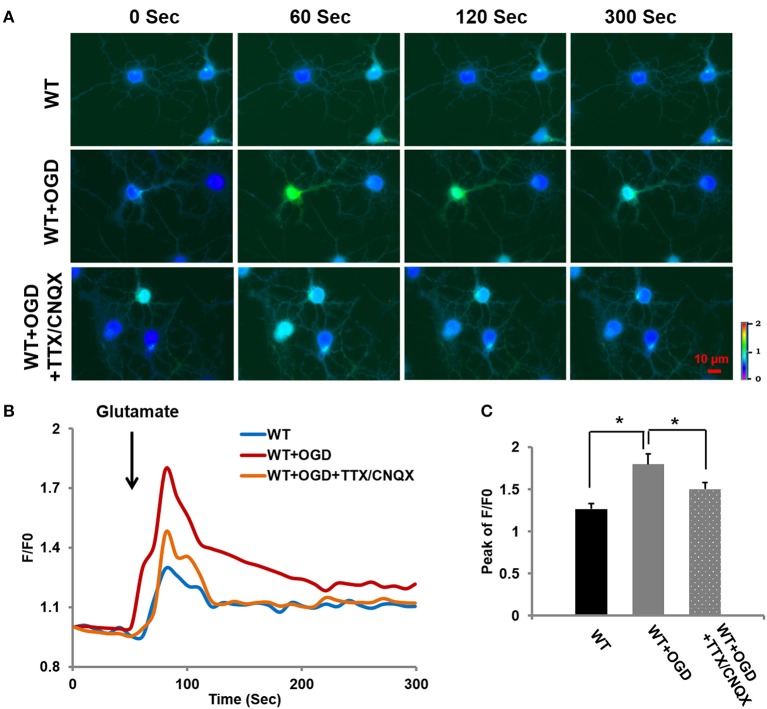
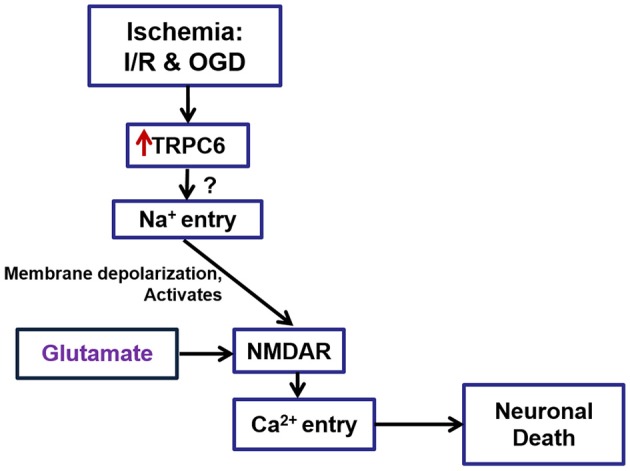
Transient receptor potential canonical 6 (TRPC6) channels are permeable to Na+ and Ca2+ and are widely expressed in the brain. In this study, the role of TRPC6 was investigated following ischemia/reperfusion (I/R) and oxygen-glucose deprivation (OGD). We found that TRPC6 expression was increased in wild-type (WT) mice cortical neurons following I/R and in primary neurons with OGD, and that deletion of TRPC6 reduced the I/R-induced brain infarct in mice and the OGD- /neurotoxin-induced neuronal death. Using live-cell imaging to examine intracellular Ca2+ levels ([Ca2+] i ), we found that OGD induced a significant higher increase in glutamate-evoked Ca2+ influx compared to untreated control and such an increase was reduced by TRPC6 deletion. Enhancement of TRPC6 expression using AdCMV-TRPC6-GFP infection in WT neurons increased [Ca2+] i in response to glutamate application compared to AdCMV-GFP control. Inhibition of N-methyl-d-aspartic acid receptor (NMDAR) with MK801 decreased TRPC6-dependent increase of [Ca2+] i in TRPC6 infected cells, indicating that such a Ca2+ influx was NMDAR dependent. Furthermore, TRPC6-dependent Ca2+ influx was blunted by blockade of Na+ entry in TRPC6 infected cells. Finally, OGD-enhanced Ca2+ influx was reduced, but not completely blocked, in the presence of voltage-dependent Na+ channel blocker tetrodotoxin (TTX) and dl-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) blocker CNQX. Altogether, we concluded that I/R-induced brain damage was, in part, due to upregulation of TRPC6 in cortical neurons. We postulate that overexpression of TRPC6 following I/R may induce neuronal death partially through TRPC6-dependent Na+ entry which activated NMDAR, thus leading to a damaging Ca2+ overload. These findings may provide a potential target for future intervention in stroke-induced brain damage.
Keywords: Ca2+; NMDA; TRPC6; ischemia; neurotoxicity; oxygen-glucose deprivation (OGD).
Figures
Similar articles
-
Effects of ion channel blockade on the distribution of Na, K, Ca and other elements in oxygen-glucose deprived CA1 hippocampal neurons.Neuroscience. 2001;103(4):971-83. doi: 10.1016/s0306-4522(01)00035-5. Neuroscience. 2001. PMID: 11301205
-
TRPC6 inhibited NMDA receptor activities and protected neurons from ischemic excitotoxicity.J Neurochem. 2012 Dec;123(6):1010-8. doi: 10.1111/jnc.12045. Epub 2012 Nov 1. J Neurochem. 2012. PMID: 23043486
-
Pharmacological inhibition of the Na(+)/Ca(2+) exchanger enhances depolarizations induced by oxygen/glucose deprivation but not responses to excitatory amino acids in rat striatal neurons.Stroke. 1999 Aug;30(8):1687-94. doi: 10.1161/01.str.30.8.1687. Stroke. 1999. PMID: 10436122
-
Role of the ubiquitin-proteasome system in brain ischemia: friend or foe?Prog Neurobiol. 2014 Jan;112:50-69. doi: 10.1016/j.pneurobio.2013.10.003. Epub 2013 Oct 22. Prog Neurobiol. 2014. PMID: 24157661 Review.
-
Novel Mechanistic Insights and Potential Therapeutic Impact of TRPC6 in Neurovascular Coupling and Ischemic Stroke.Int J Mol Sci. 2021 Feb 19;22(4):2074. doi: 10.3390/ijms22042074. Int J Mol Sci. 2021. PMID: 33669830 Free PMC article. Review.
Cited by
-
Neuroprotection by cattle encephalon glycoside and ignotin beyond the time window of thrombolysis in ischemic stroke.Neural Regen Res. 2021 Feb;16(2):312-318. doi: 10.4103/1673-5374.290899. Neural Regen Res. 2021. PMID: 32859790 Free PMC article.
-
2-(2-Benzofuranyl)-2-imidazoline treatment within 5 hours after cerebral ischemia/reperfusion protects the brain.Neural Regen Res. 2018 Dec;13(12):2111-2118. doi: 10.4103/1673-5374.241461. Neural Regen Res. 2018. PMID: 30323139 Free PMC article.
-
Novel Targets for Stroke Therapy: Special Focus on TRPC Channels and TRPC6.Front Aging Neurosci. 2020 Mar 18;12:70. doi: 10.3389/fnagi.2020.00070. eCollection 2020. Front Aging Neurosci. 2020. PMID: 32256338 Free PMC article. Review.
-
Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications.Front Neurosci. 2020 Oct 6;14:572965. doi: 10.3389/fnins.2020.572965. eCollection 2020. Front Neurosci. 2020. PMID: 33117120 Free PMC article. Review.
-
Propofol inhibited autophagy through Ca2+/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury.Mol Med. 2018 Nov 23;24(1):58. doi: 10.1186/s10020-018-0054-1. Mol Med. 2018. PMID: 30470173 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous