

Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury
- PMID: 28401475
- PMCID: PMC5636654
- DOI: 10.1007/s12035-017-0503-9
Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury
Abstract
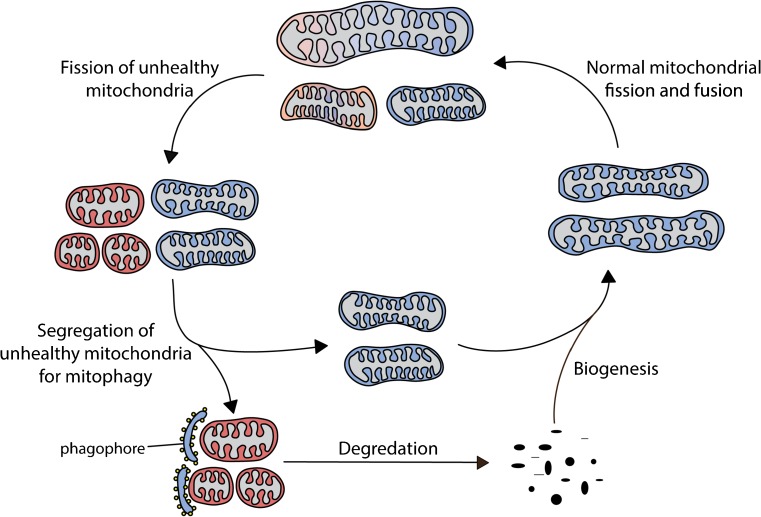
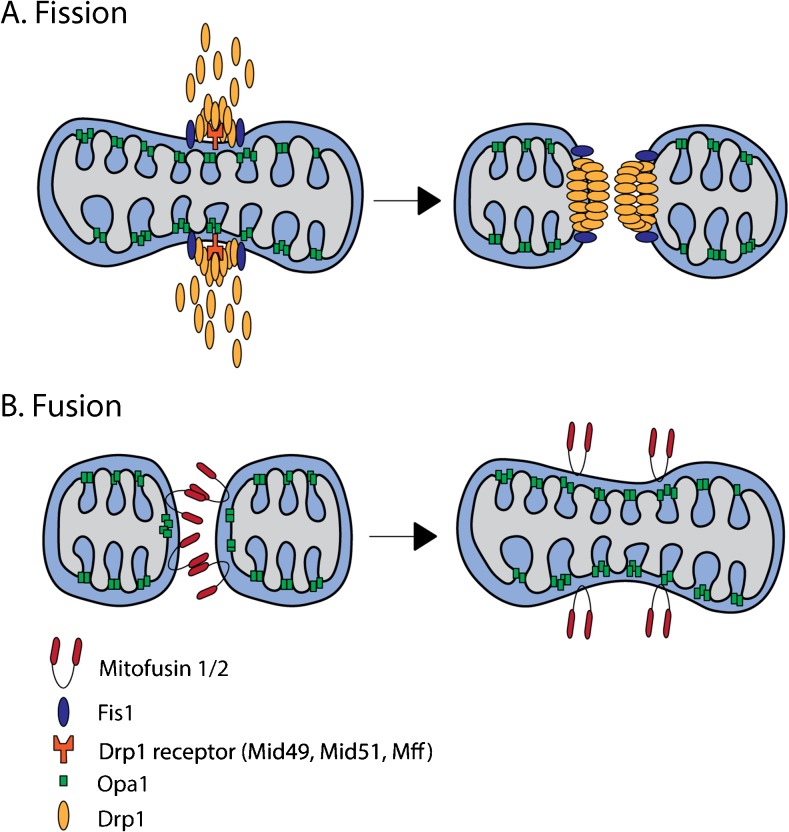
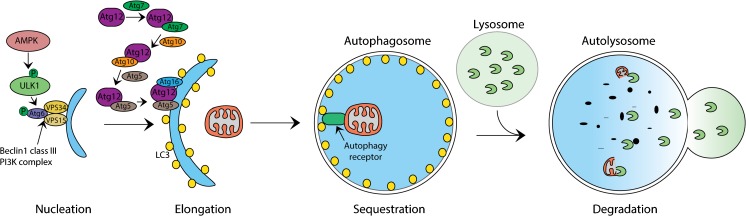
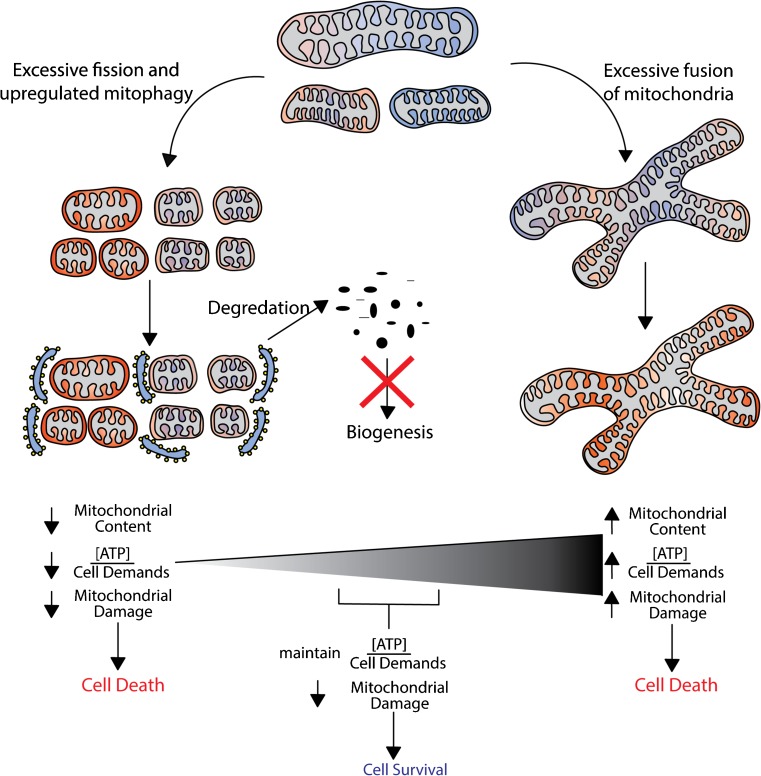
Mitochondria are key regulators of cell fate during disease. They control cell survival via the production of ATP that fuels cellular processes and, conversely, cell death via the induction of apoptosis through release of pro-apoptotic factors such as cytochrome C. Therefore, it is essential to have stringent quality control mechanisms to ensure a healthy mitochondrial network. Quality control mechanisms are largely regulated by mitochondrial dynamics and mitophagy. The processes of mitochondrial fission (division) and fusion allow for damaged mitochondria to be segregated and facilitate the equilibration of mitochondrial components such as DNA, proteins, and metabolites. The process of mitophagy are responsible for the degradation and recycling of damaged mitochondria. These mitochondrial quality control mechanisms have been well studied in chronic and acute pathologies such as Parkinson's disease, Alzheimer's disease, stroke, and acute myocardial infarction, but less is known about how these two processes interact and contribute to specific pathophysiologic states. To date, evidence for the role of mitochondrial quality control in acute and chronic disease is divergent and suggests that mitochondrial quality control processes can serve both survival and death functions depending on the disease state. This review aims to provide a synopsis of the molecular mechanisms involved in mitochondrial quality control, to summarize our current understanding of the complex role that mitochondrial quality control plays in the progression of acute vs chronic diseases and, finally, to speculate on the possibility that targeted manipulation of mitochondrial quality control mechanisms may be exploited for the rationale design of novel therapeutic interventions.
Keywords: Brain; Ischemia; Mitochondria; Mitochondrial dynamics; Mitophagy; Reperfusion.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
