

STIM1 Ca2+ Sensor Control of L-type Ca2+-Channel-Dependent Dendritic Spine Structural Plasticity and Nuclear Signaling
- PMID: 28402855
- PMCID: PMC5451256
- DOI: 10.1016/j.celrep.2017.03.056
STIM1 Ca2+ Sensor Control of L-type Ca2+-Channel-Dependent Dendritic Spine Structural Plasticity and Nuclear Signaling
Abstract
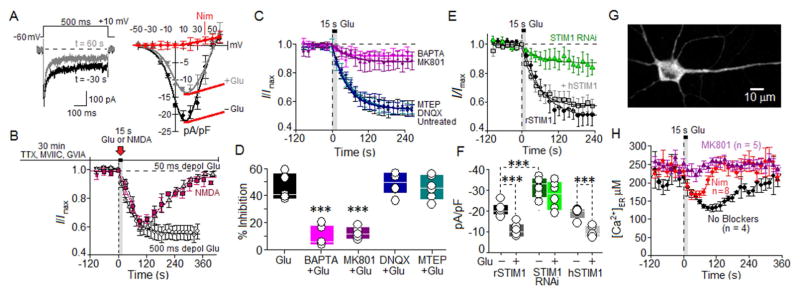
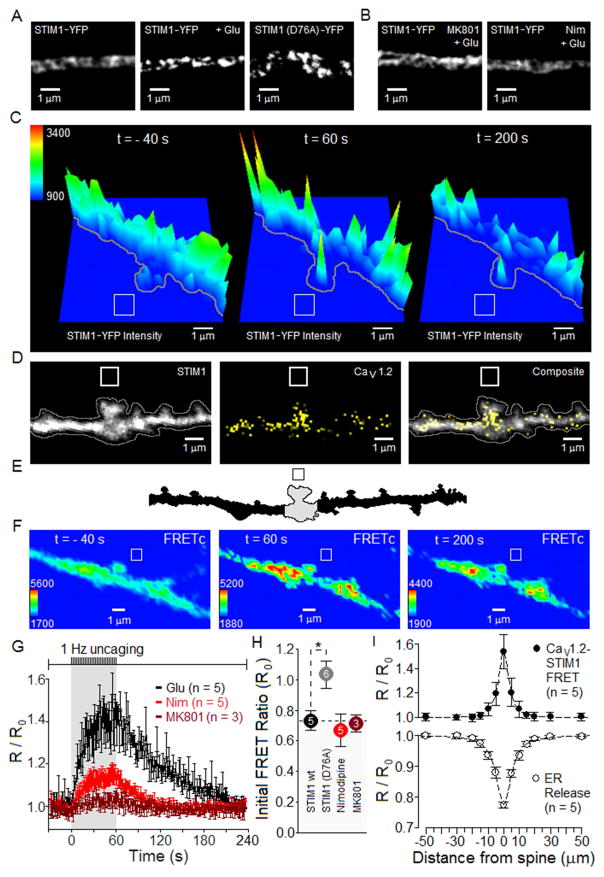
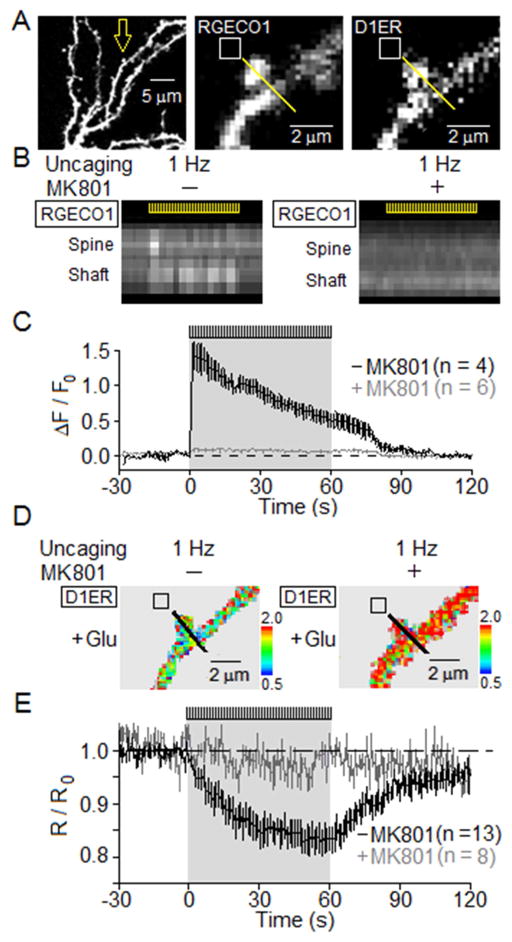
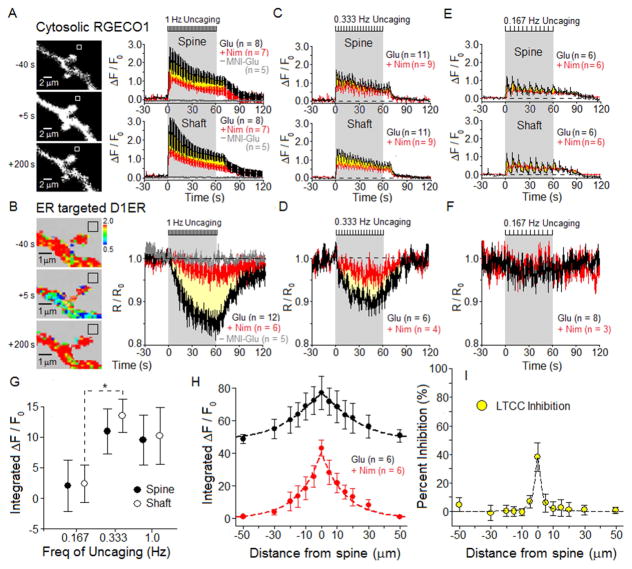
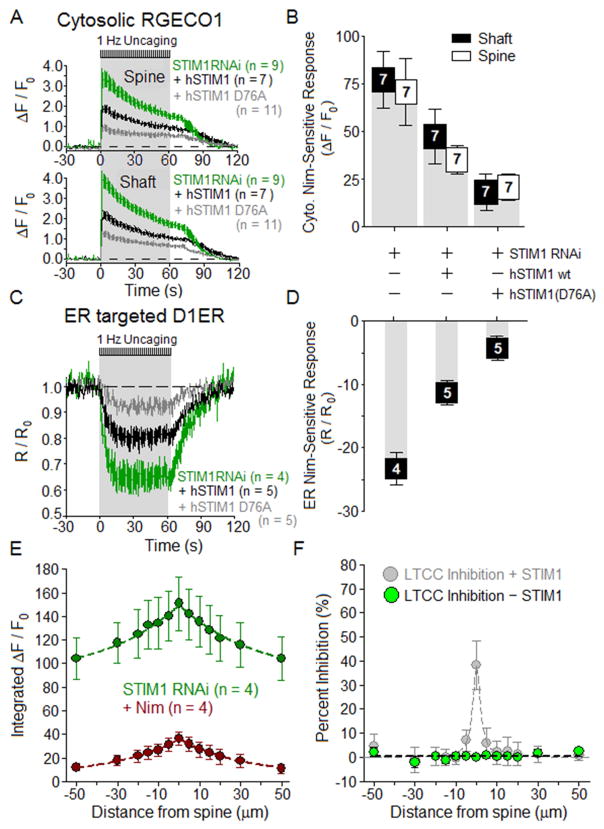
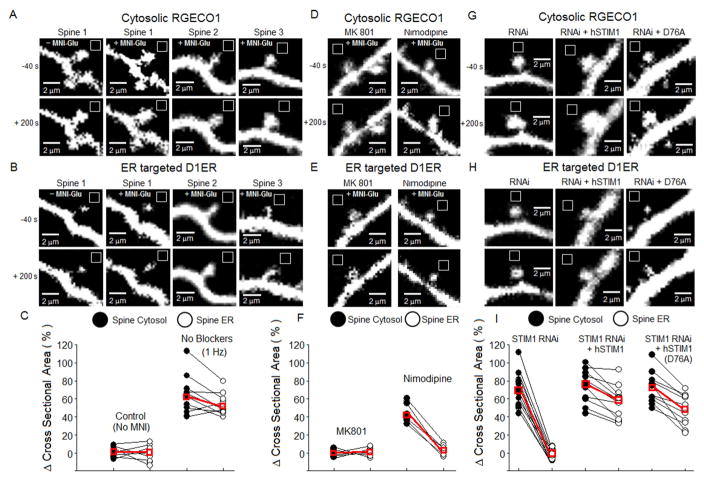
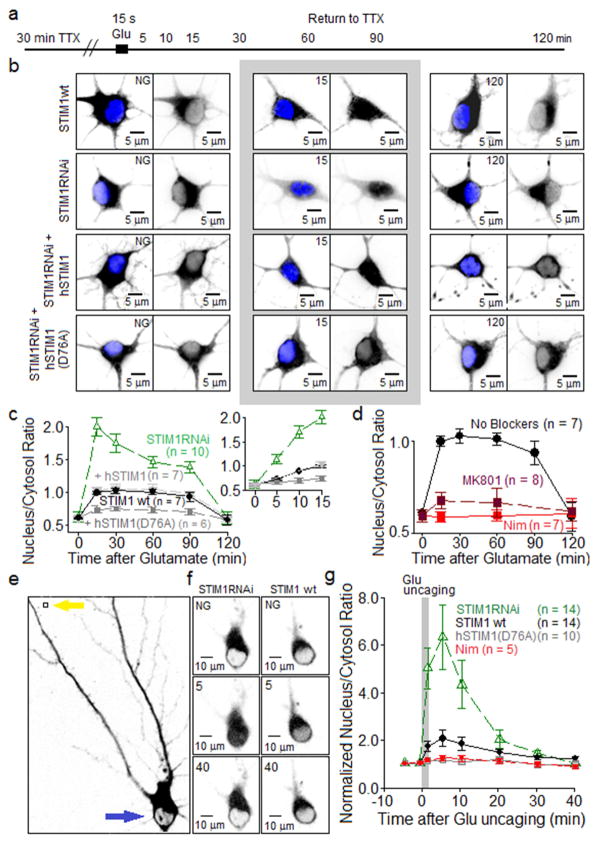
Potentiation of synaptic strength relies on postsynaptic Ca2+ signals, modification of dendritic spine structure, and changes in gene expression. One Ca2+ signaling pathway supporting these processes routes through L-type Ca2+ channels (LTCC), whose activity is subject to tuning by multiple mechanisms. Here, we show in hippocampal neurons that LTCC inhibition by the endoplasmic reticulum (ER) Ca2+ sensor, stromal interaction molecule 1 (STIM1), is engaged by the neurotransmitter glutamate, resulting in regulation of spine ER structure and nuclear signaling by the NFATc3 transcription factor. In this mechanism, depolarization by glutamate activates LTCC Ca2+ influx, releases Ca2+ from the ER, and consequently drives STIM1 aggregation and an inhibitory interaction with LTCCs that increases spine ER content but decreases NFATc3 nuclear translocation. These findings of negative feedback control of LTCC signaling by STIM1 reveal interplay between Ca2+ influx and release from stores that controls both postsynaptic structural plasticity and downstream nuclear signaling.
Keywords: L-type Ca(2+) channel; N-methyl-D-aspartate receptor; cytoplasmic Ca(2+); dendritic spine; endoplasmic reticulum; glutamate; nuclear factor of activated T cells; stromal interaction molecule 1; structural plasticity; voltage-gated Ca(2+) channel.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
