

Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency
- PMID: 28406489
- PMCID: PMC5589980
- DOI: 10.1038/gim.2017.7
Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency
Abstract
Disclaimer:This diagnostic guideline is intended as an educational resource and represents the opinions of the authors, and is not representative of recommendations or policy of the American College of Medical Genetics and Genomics (ACMG). The information should be considered a consensus based on expert opinion, as more comprehensive levels of evidence were not available in the literature in all cases.
Background: Acid sphingomyelinase deficiency (ASMD) is a rare, progressive, and often fatal lysosomal storage disease. The underlying metabolic defect is deficiency of the enzyme acid sphingomyelinase that results in progressive accumulation of sphingomyelin in target tissues. ASMD manifests as a spectrum of severity ranging from rapidly progressive severe neurovisceral disease that is uniformly fatal to more slowly progressive chronic neurovisceral and chronic visceral forms. Disease management is aimed at symptom control and regular assessments for multisystem involvement.
Purpose and methods: An international panel of experts in the clinical and laboratory evaluation, diagnosis, treatment/management, and genetic aspects of ASMD convened to review the evidence base and share personal experience in order to develop a guideline for diagnosis of the various ASMD phenotypes.
Conclusions: Although care of ASMD patients is typically provided by metabolic disease specialists, the guideline is directed at a wide range of providers because it is important for primary care providers (e.g., pediatricians and internists) and specialists (e.g., pulmonologists, hepatologists, and hematologists) to be able to identify ASMD.Genet Med advance online publication 13 April 2017.
Figures
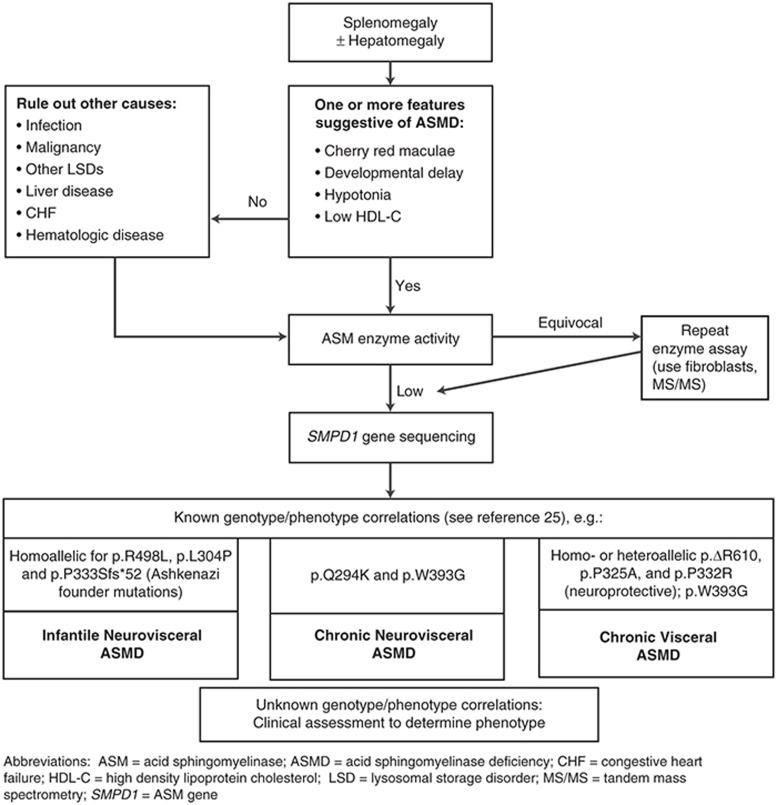
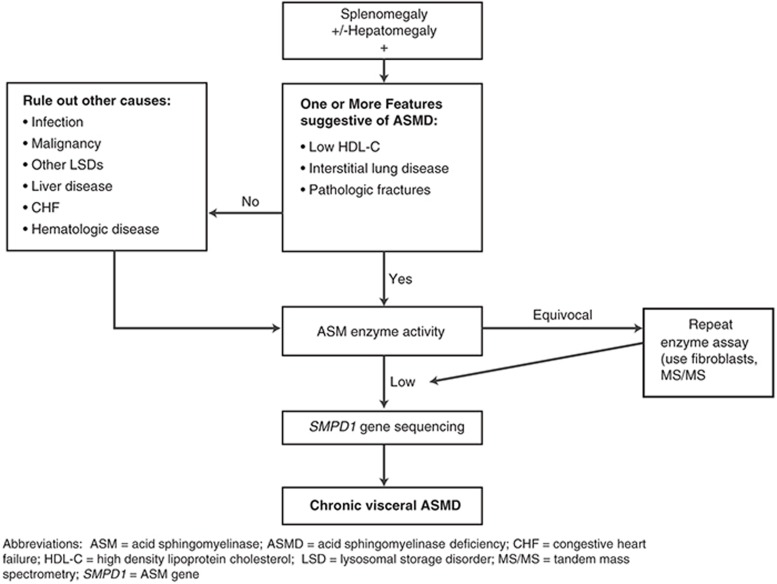
References
-
- Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. In: Valle D, Beaudet A, Vogelstein B, et al. (eds). OMMBID—The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill: New York, 2013. http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62643414. Accessed January 2015.
-
- Kingma SD, Bodamer OA, Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab 2015;29:145–157. - PubMed
-
- Schuchman EH, Wasserstein MP. Types A and B Niemann-Pick disease. Best Pract Res Clin Endocrinol Metab 2015;29:237–247. - PubMed
-
- McGovern MM, Aron A, Brodie SE, Desnick RJ, Wasserstein MP. Natural history of Type A Niemann-Pick disease: possible endpoints for therapeutic trials. Neurology 2006;66:228–232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
