

Gene co-expression network connectivity is an important determinant of selective constraint
- PMID: 28406900
- PMCID: PMC5407845
- DOI: 10.1371/journal.pgen.1006402
Gene co-expression network connectivity is an important determinant of selective constraint
Abstract
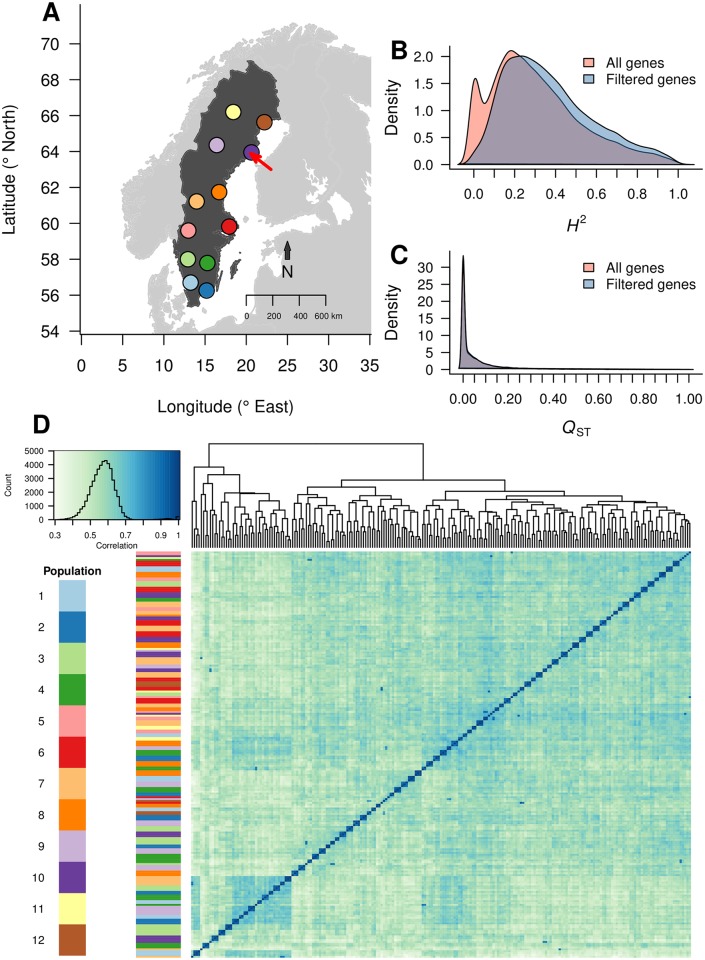
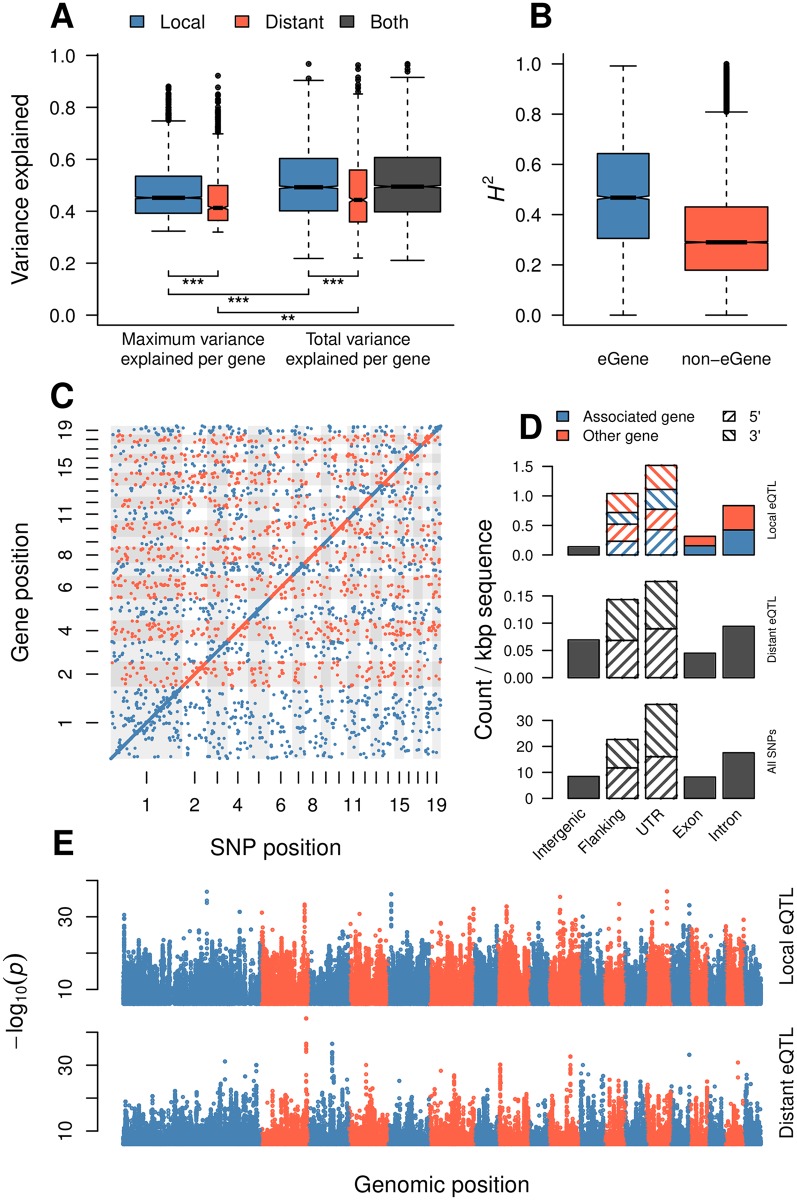
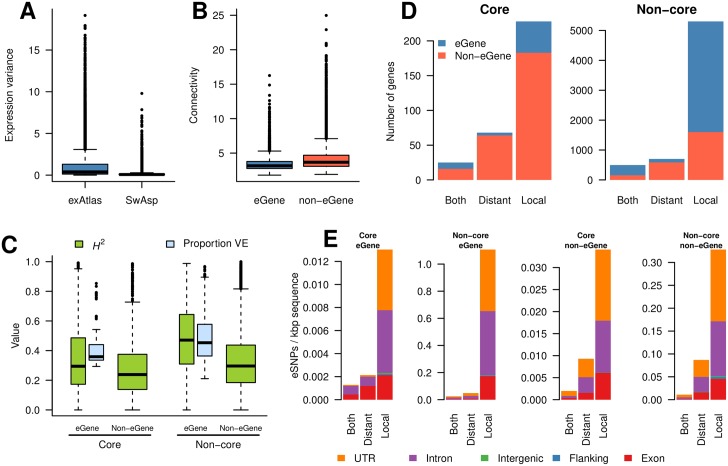
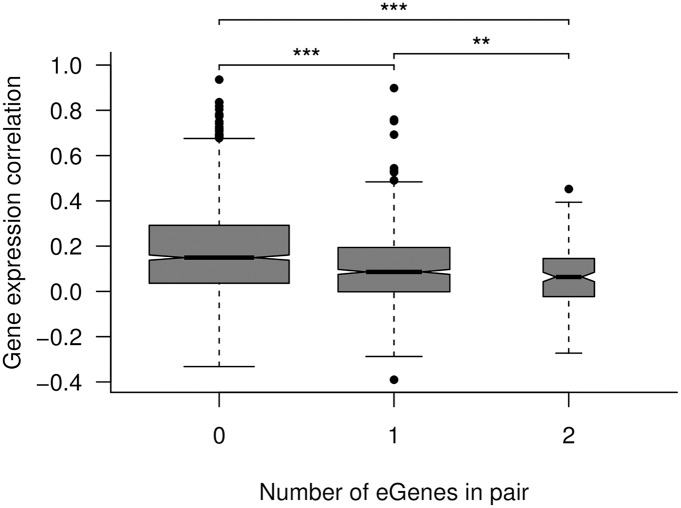
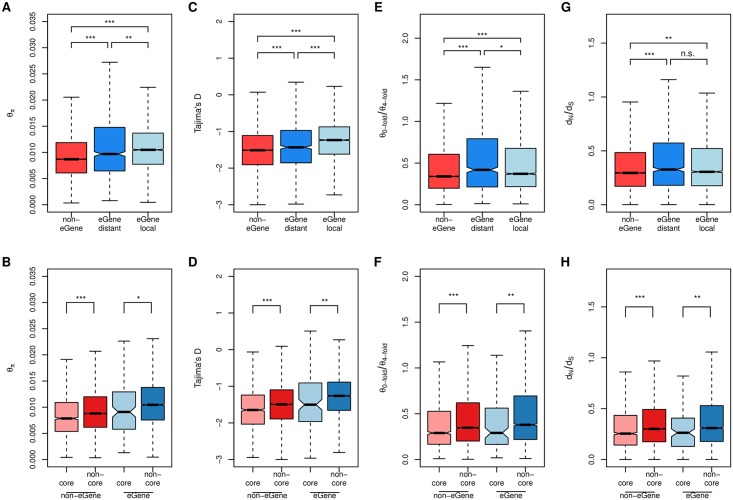
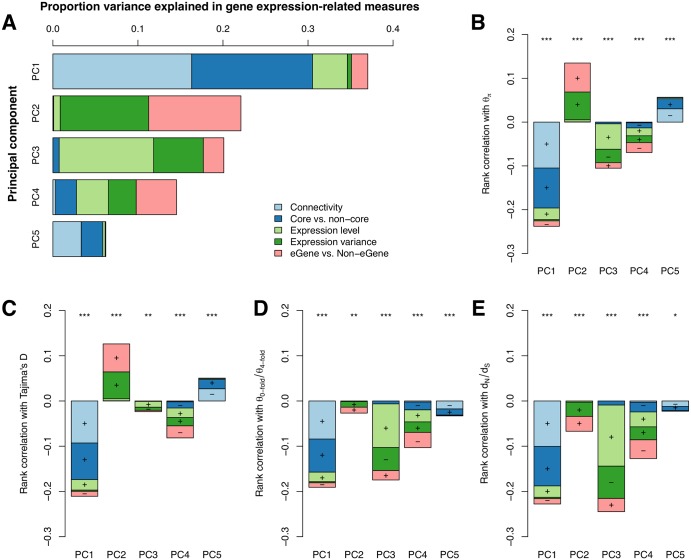
While several studies have investigated general properties of the genetic architecture of natural variation in gene expression, few of these have considered natural, outbreeding populations. In parallel, systems biology has established that a general feature of biological networks is that they are scale-free, rendering them buffered against random mutations. To date, few studies have attempted to examine the relationship between the selective processes acting to maintain natural variation of gene expression and the associated co-expression network structure. Here we utilised RNA-Sequencing to assay gene expression in winter buds undergoing bud flush in a natural population of Populus tremula, an outbreeding forest tree species. We performed expression Quantitative Trait Locus (eQTL) mapping and identified 164,290 significant eQTLs associating 6,241 unique genes (eGenes) with 147,419 unique SNPs (eSNPs). We found approximately four times as many local as distant eQTLs, with local eQTLs having significantly higher effect sizes. eQTLs were primarily located in regulatory regions of genes (UTRs or flanking regions), regardless of whether they were local or distant. We used the gene expression data to infer a co-expression network and investigated the relationship between network topology, the genetic architecture of gene expression and signatures of selection. Within the co-expression network, eGenes were underrepresented in network module cores (hubs) and overrepresented in the periphery of the network, with a negative correlation between eQTL effect size and network connectivity. We additionally found that module core genes have experienced stronger selective constraint on coding and non-coding sequence, with connectivity associated with signatures of selection. Our integrated genetics and genomics results suggest that purifying selection is the primary mechanism underlying the genetic architecture of natural variation in gene expression assayed in flushing leaf buds of P. tremula and that connectivity within the co-expression network is linked to the strength of purifying selection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Ingvarsson PK, Hvidsten TR, Street NR. Towards integration of population and comparative genomics in forest trees. New Phytol. 2016; - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
