

Segmentum: a tool for copy number analysis of cancer genomes
- PMID: 28407731
- PMCID: PMC5390478
- DOI: 10.1186/s12859-017-1626-8
Segmentum: a tool for copy number analysis of cancer genomes
Abstract
Background: Somatic alterations, including loss of heterozygosity, can affect the expression of oncogenes and tumor suppressor genes. Whole genome sequencing enables detailed characterization of such aberrations. However, due to the limitations of current high throughput sequencing technologies, this task remains challenging. Hence, accurate and reliable detection of such events is crucial for the identification of cancer-related alterations.
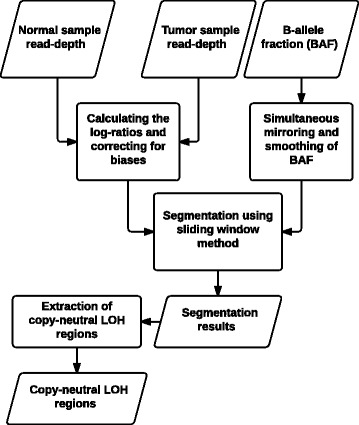
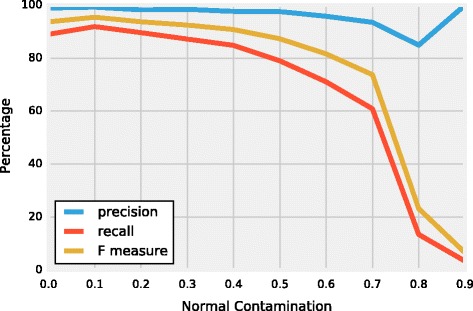
Results: We introduce a new tool called Segmentum for determining somatic copy numbers using whole genome sequencing from paired tumor/normal samples. In our approach, read depth and B-allele fraction signals are smoothed, and double sliding windows are used to detect breakpoints, which makes our approach fast and straightforward. Because the breakpoint detection is performed simultaneously at different scales, it allows accurate detection as suggested by the evaluation results from simulated and real data. We applied Segmentum to paired tumor/normal whole genome sequencing samples from 38 patients with low-grade glioma from the TCGA dataset and were able to confirm the recurrence of copy-neutral loss of heterozygosity in chromosome 17p in low-grade astrocytoma characterized by IDH1/2 mutation and lack of 1p/19q co-deletion, which was previously reported using SNP array data.
Conclusions: Segmentum is an accurate, user-friendly tool for somatic copy number analysis of tumor samples. We demonstrate that this tool is suitable for the analysis of large cohorts, such as the TCGA dataset.
Keywords: Cancer; Loss of heterozygosity; Segmentation; Somatic copy number analysis; Whole-genome sequencing.
Figures




References
-
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. - DOI - PMC - PubMed
-
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, METABRIC Group. Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. - PMC - PubMed
-
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, Chin SF, Turashvili G, Hirst M, Caldas C, Marra MA, Aparicio S, Shah SP. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22(10):1995–2007. doi: 10.1101/gr.137570.112. - DOI - PMC - PubMed
-
- Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, Yamamoto T, Tanahashi K, Ranjit M, Wakabayashi T, Yoshizato T, Kataoka K, Yoshida K, Nagata Y, Sato-Otsubo A, Tanaka H, Sanada M, Kondo Y, Nakamura H, Mizoguchi M, Abe T, Muragaki Y, Watanabe R, Ito I, Miyano S, Natsume A, Ogawa S. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47(5):458–468. doi: 10.1038/ng.3273. - DOI - PubMed
-
- Barresi V, Romano A, Musso N, Capizzi C, Consoli C, Martelli MP, Palumbo G, Di Raimondo F, Condorelli DF. Broad copy neutral-loss of heterozygosity regions and rare recurring copy number abnormalities in normal karyotype-acute myeloid leukemia genomes. Genes Chromosomes Cancer. 2010;49(11):1014–1023. doi: 10.1002/gcc.20810. - DOI - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
