

Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non-uniformity
- PMID: 28408746
- PMCID: PMC5429826
- DOI: 10.1038/s41598-017-01005-x
Novel metrics to measure coverage in whole exome sequencing datasets reveal local and global non-uniformity
Abstract
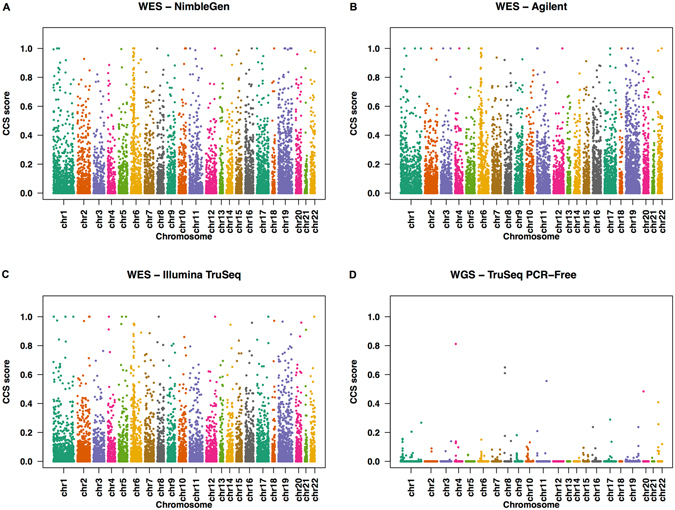
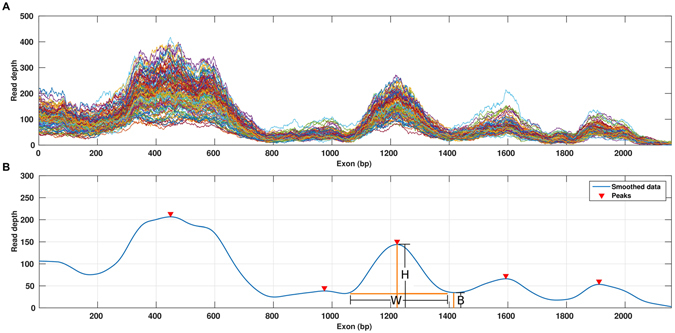
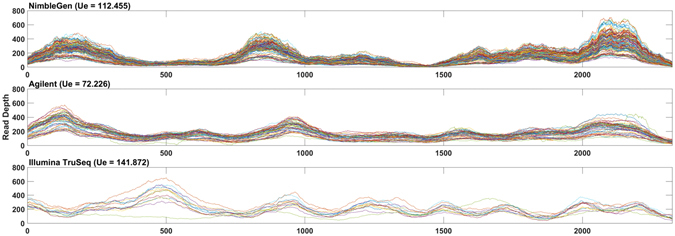
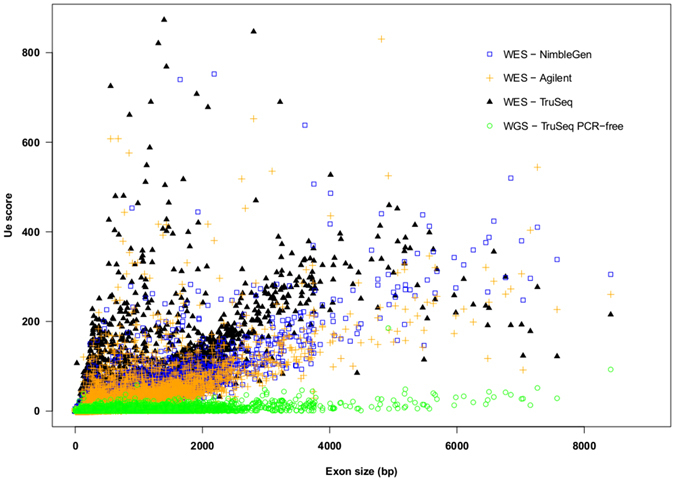
Whole Exome Sequencing (WES) is a powerful clinical diagnostic tool for discovering the genetic basis of many diseases. A major shortcoming of WES is uneven coverage of sequence reads over the exome targets contributing to many low coverage regions, which hinders accurate variant calling. In this study, we devised two novel metrics, Cohort Coverage Sparseness (CCS) and Unevenness (UE) Scores for a detailed assessment of the distribution of coverage of sequence reads. Employing these metrics we revealed non-uniformity of coverage and low coverage regions in the WES data generated by three different platforms. This non-uniformity of coverage is both local (coverage of a given exon across different platforms) and global (coverage of all exons across the genome in the given platform). The low coverage regions encompassing functionally important genes were often associated with high GC content, repeat elements and segmental duplications. While a majority of the problems associated with WES are due to the limitations of the capture methods, further refinements in WES technologies have the potential to enhance its clinical applications.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
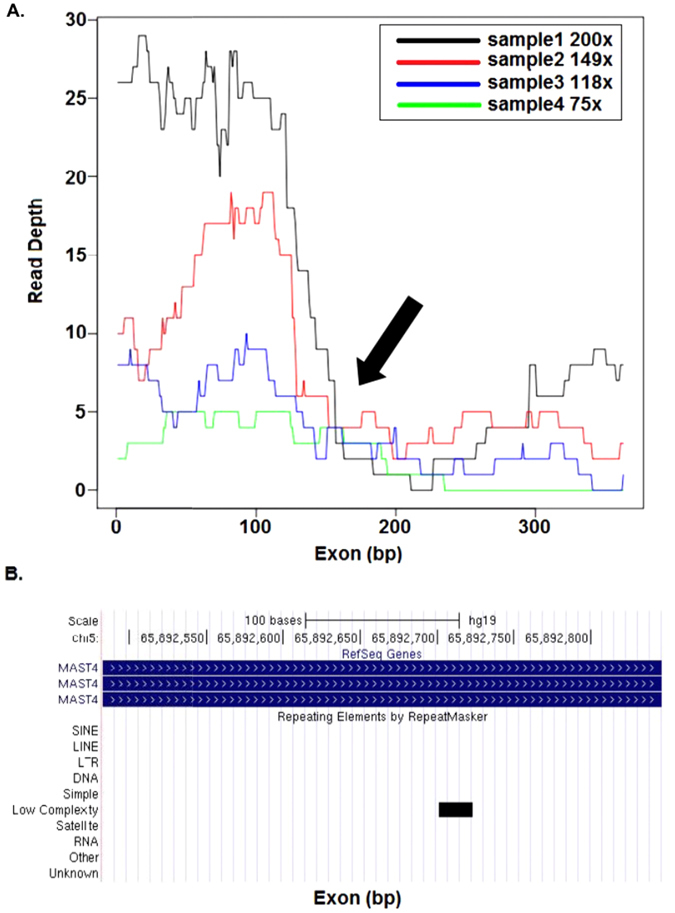
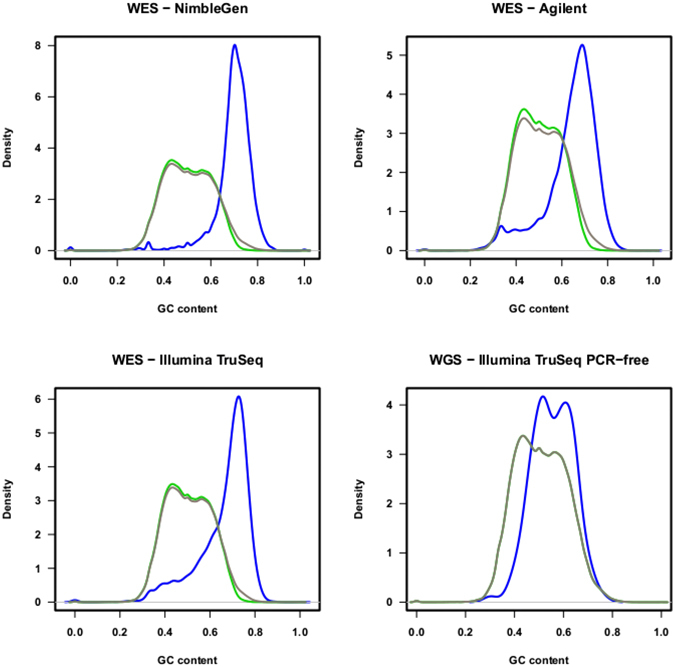
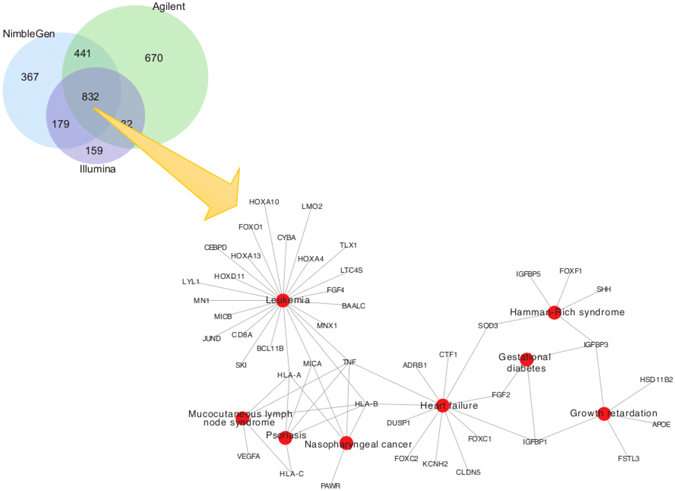
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
