

Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis
- PMID: 28409282
- PMCID: PMC5427160
- DOI: 10.1007/s00401-017-1708-8
Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis
Abstract
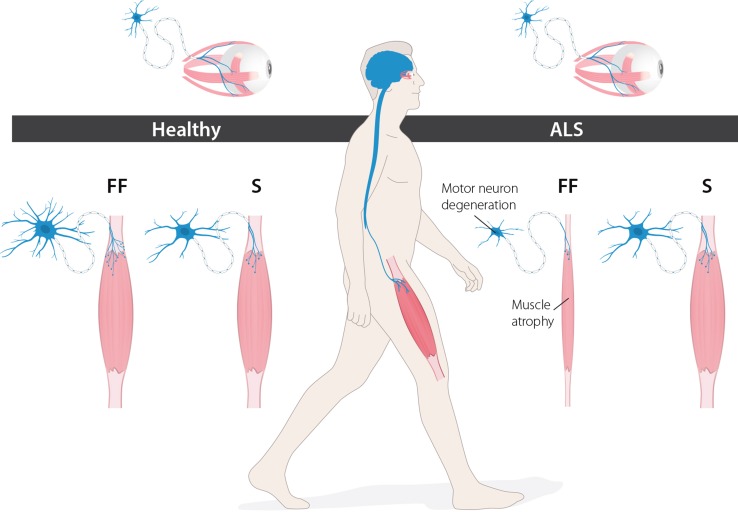
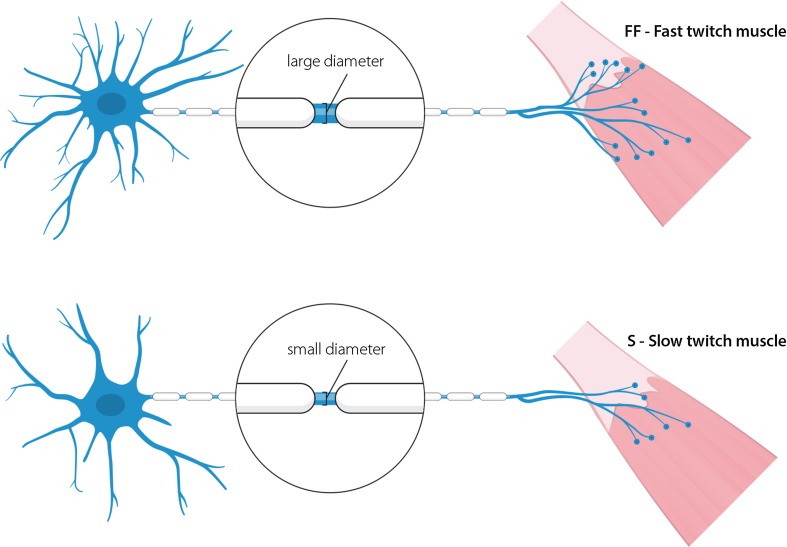
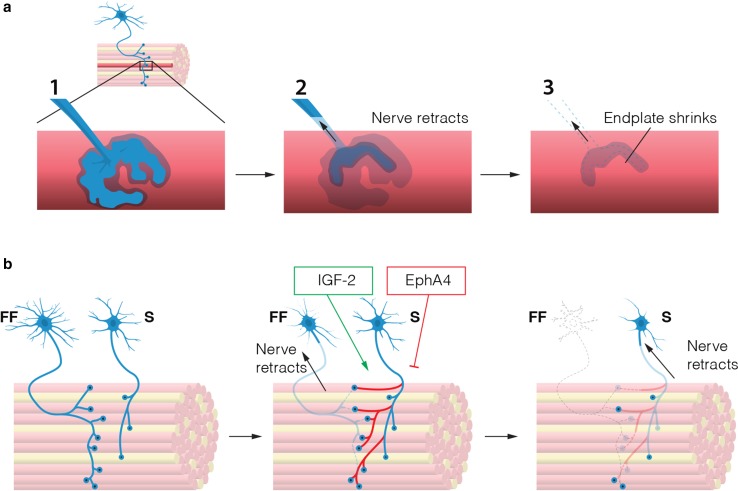
In the fatal disease-amyotrophic lateral sclerosis (ALS)-upper (corticospinal) motor neurons (MNs) and lower somatic MNs, which innervate voluntary muscles, degenerate. Importantly, certain lower MN subgroups are relatively resistant to degeneration, even though pathogenic proteins are typically ubiquitously expressed. Ocular MNs (OMNs), including the oculomotor, trochlear and abducens nuclei (CNIII, IV and VI), which regulate eye movement, persist throughout the disease. Consequently, eye-tracking devices are used to enable paralysed ALS patients (who can no longer speak) to communicate. Additionally, there is a gradient of vulnerability among spinal MNs. Those innervating fast-twitch muscle are most severely affected and degenerate first. MNs innervating slow-twitch muscle can compensate temporarily for the loss of their neighbours by re-innervating denervated muscle until later in disease these too degenerate. The resistant OMNs and the associated extraocular muscles (EOMs) are anatomically and functionally very different from other motor units. The EOMs have a unique set of myosin heavy chains, placing them outside the classical characterization spectrum of all skeletal muscle. Moreover, EOMs have multiple neuromuscular innervation sites per single myofibre. Spinal fast and slow motor units show differences in their dendritic arborisations and the number of myofibres they innervate. These motor units also differ in their functionality and excitability. Identifying the molecular basis of cell-intrinsic pathways that are differentially activated between resistant and vulnerable MNs could reveal mechanisms of selective neuronal resistance, degeneration and regeneration and lead to therapies preventing progressive MN loss in ALS. Illustrating this, overexpression of OMN-enriched genes in spinal MNs, as well as suppression of fast spinal MN-enriched genes can increase the lifespan of ALS mice. Here, we discuss the pattern of lower MN degeneration in ALS and review the current literature on OMN resistance in ALS and differential spinal MN vulnerability. We also reflect upon the non-cell autonomous components that are involved in lower MN degeneration in ALS.
Keywords: ALS; Fast and slow motor units; Neurodegeneration; Neuromuscular junction; Oculomotor neuron; Selective vulnerability.
Figures




References
-
- Al-Chalabi A, Andersen PM, Chioza B, Shaw C, Sham PC, Robberecht W, Matthijs G, Camu W, Marklund SL, Forsgren L. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet. 1998;7:2045–2050. doi: 10.1093/hmg/7.13.2045. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
