

Inflammation and Fibrosis in Polycystic Kidney Disease
- PMID: 28409351
- PMCID: PMC7875307
- DOI: 10.1007/978-3-319-51436-9_12
Inflammation and Fibrosis in Polycystic Kidney Disease
Abstract
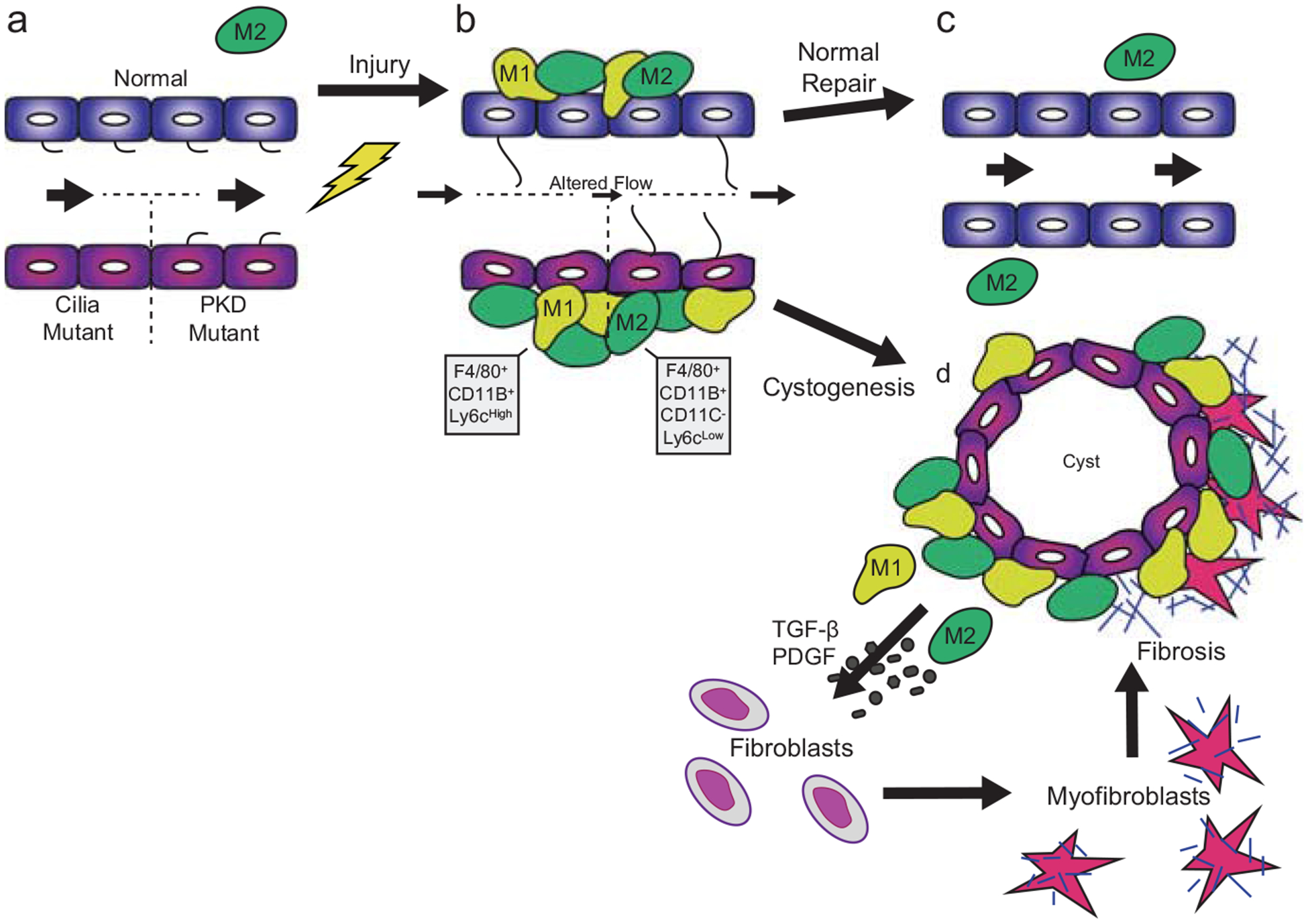
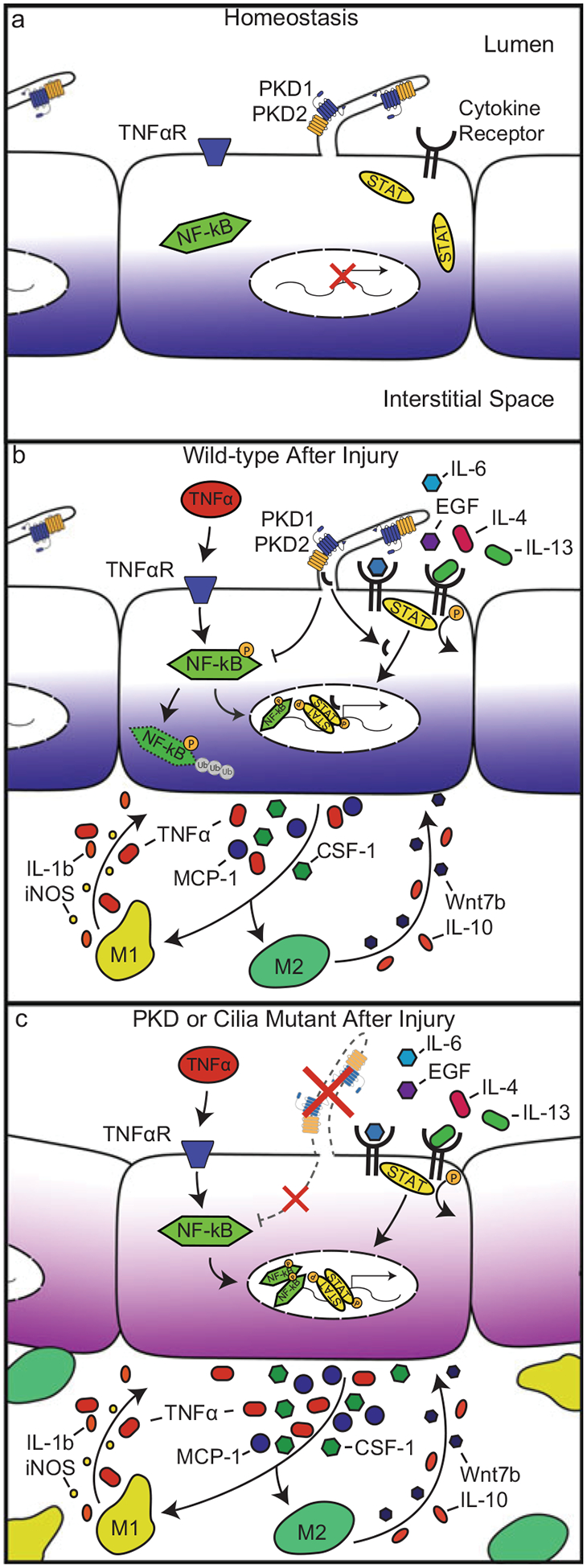
Polycystic kidney disease (PKD) is a commonly inherited disorder characterized by cyst formation and fibrosis (Wilson, N Engl J Med 350:151-164, 2004) and is caused by mutations in cilia or cilia-related proteins, such as polycystin 1 or 2 (Oh and Katsanis, Development 139:443-448, 2012; Kotsis et al., Nephrol Dial Transplant 28:518-526, 2013). A major pathological feature of PKD is the development of interstitial inflammation and fibrosis with an associated accumulation of inflammatory cells (Grantham, N Engl J Med 359:1477-1485, 2008; Zeier et al., Kidney Int 42:1259-1265, 1992; Ibrahim, Sci World J 7:1757-1767, 2007). It is unclear whether inflammation is a driving force for cyst formation or a consequence of the pathology (Ta et al., Nephrology 18:317-330, 2013) as in some murine models cysts are present prior to the increase in inflammatory cells (Phillips et al., Kidney Blood Press Res 30:129-144, 2007; Takahashi et al., J Am Soc Nephrol JASN 1:980-989, 1991), while in other models the increase in inflammatory cells is present prior to or coincident with cyst initiation (Cowley et al., Kidney Int 43:522-534, 1993, Kidney Int 60:2087-2096, 2001). Additional support for inflammation as an important contributor to cystic kidney disease is the increased expression of many pro-inflammatory cytokines in murine models and human patients with cystic kidney disease (Karihaloo et al., J Am Soc Nephrol JASN 22:1809-1814, 2011; Swenson-Fields et al., Kidney Int, 2013; Li et al., Nat Med 14:863-868, 2008a). Based on these data, an emerging model in the field is that disruption of primary cilia on tubule epithelial cells leads to abnormal cytokine cross talk between the epithelium and the inflammatory cells contributing to cyst growth and fibrosis (Ta et al., Nephrology 18:317-330, 2013). These cytokines are produced by interstitial fibroblasts, inflammatory cells, and tubule epithelial cells and activate multiple pathways including the JAK-STAT and NF-κB signaling (Qin et al., J Am Soc Nephrol JASN 23:1309-1318, 2012; Park et al., Am J Nephrol 32:169-178, 2010; Bhunia et al., Cell 109:157-168, 2002). Indeed, inflammatory cells are responsible for producing several of the pro-fibrotic growth factors observed in PKD patients with fibrosis (Nakamura et al., Am J Nephrol 20:32-36, 2000; Wilson et al., J Cell Physiol 150:360-369, 1992; Song et al., Hum Mol Genet 18:2328-2343, 2009; Schieren et al., Nephrol Dial Transplant 21:1816-1824, 2006). These growth factors trigger epithelial cell proliferation and myofibroblast activation that stimulate the production of extracellular matrix (ECM) genes including collagen types 1 and 3 and fibronectin, leading to reduced glomerular function with approximately 50% of ADPKD patients progressing to end-stage renal disease (ESRD). Therefore, treatments designed to reduce inflammation and slow the rate of fibrosis are becoming important targets that hold promise to improve patient life span and quality of life. In fact, recent studies in several PKD mouse models indicate that depletion of macrophages reduces cyst severity. In this chapter, we review the potential mechanisms of interstitial inflammation in PKD with a focus on ADPKD and discuss the role of interstitial inflammation in progression to fibrosis and ESRD.
Figures
References
-
- Adams DO, Hamilton TA (1984) The cell biology of macrophage activation. Annu Rev Immunol 2:283–318 - PubMed
-
- Albaqumi M, Srivastava S, Li Z, Zhdnova O, Wulff H, Itani O, Wallace DP, Skolnik EY (2008) KCa3.1 potassium channels are critical for cAMP-dependent chloride secretion and cyst growth in autosomal-dominant polycystic kidney disease. Kidney Int 74:740–749 - PubMed
-
- Benoit M, Desnues B, Mege JL (2008) Macrophage polarization in bacterial infections. J Immunol 181:3733–3739 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
