

RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection
- PMID: 28410401
- PMCID: PMC5406035
- DOI: 10.1371/journal.ppat.1006326
RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection
Abstract
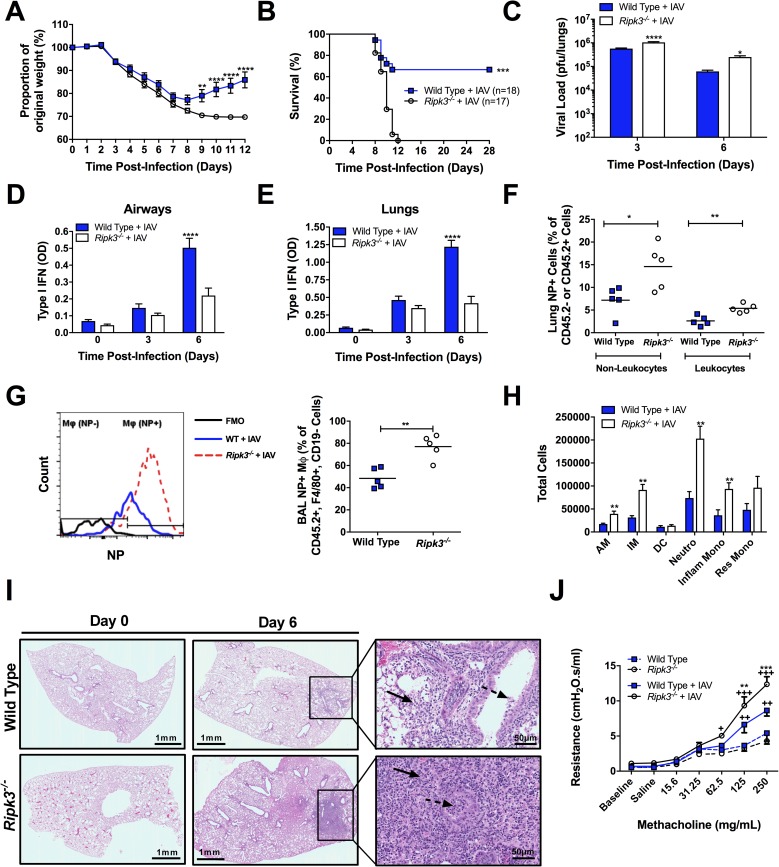
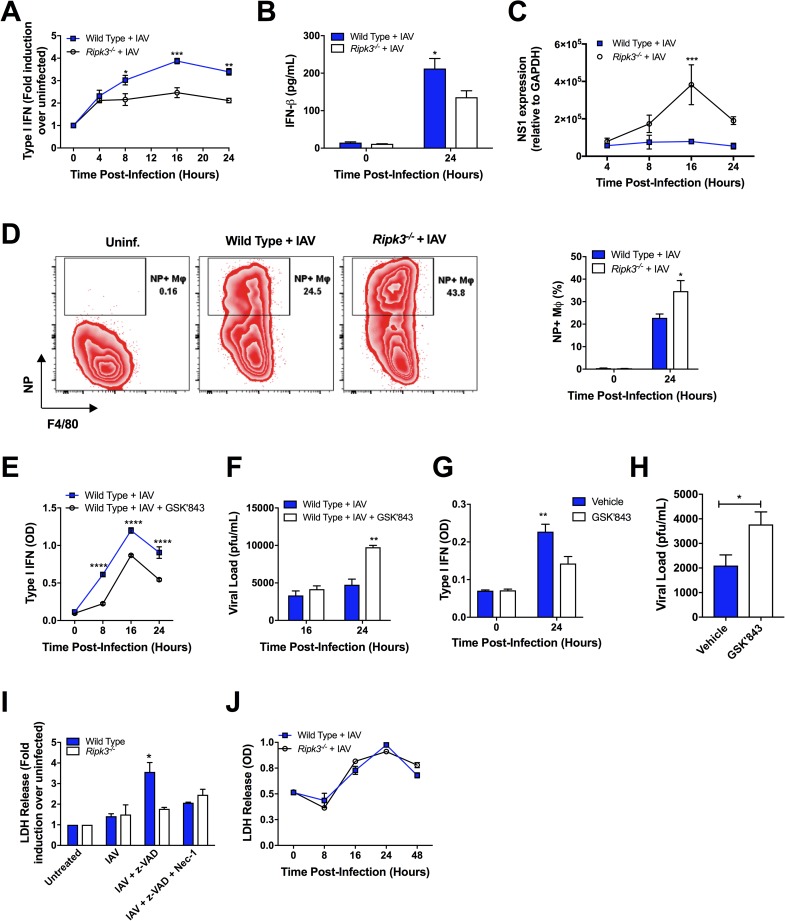
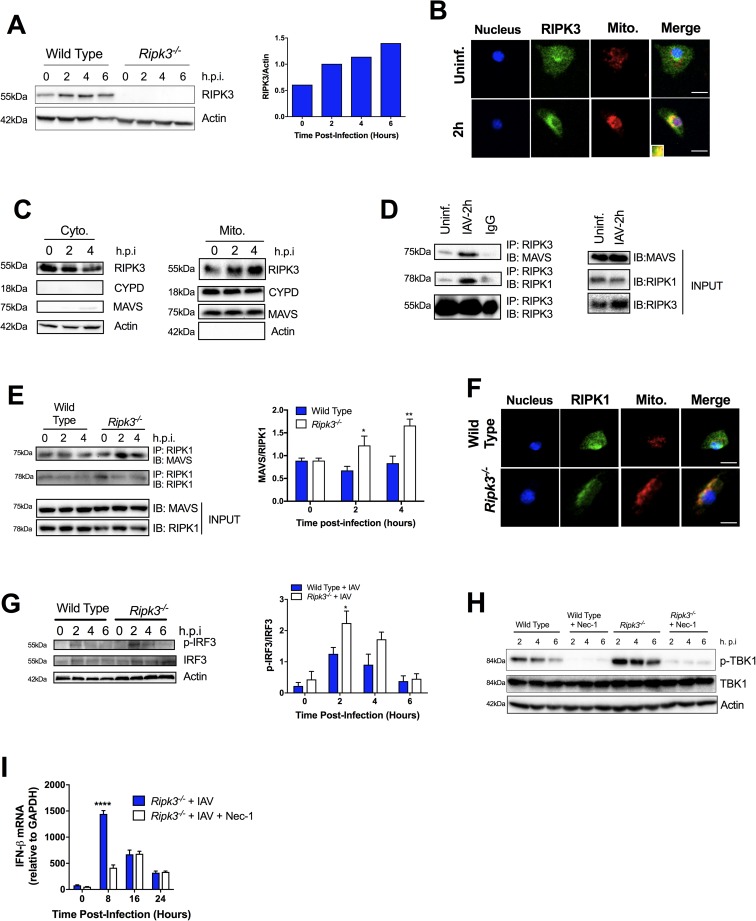
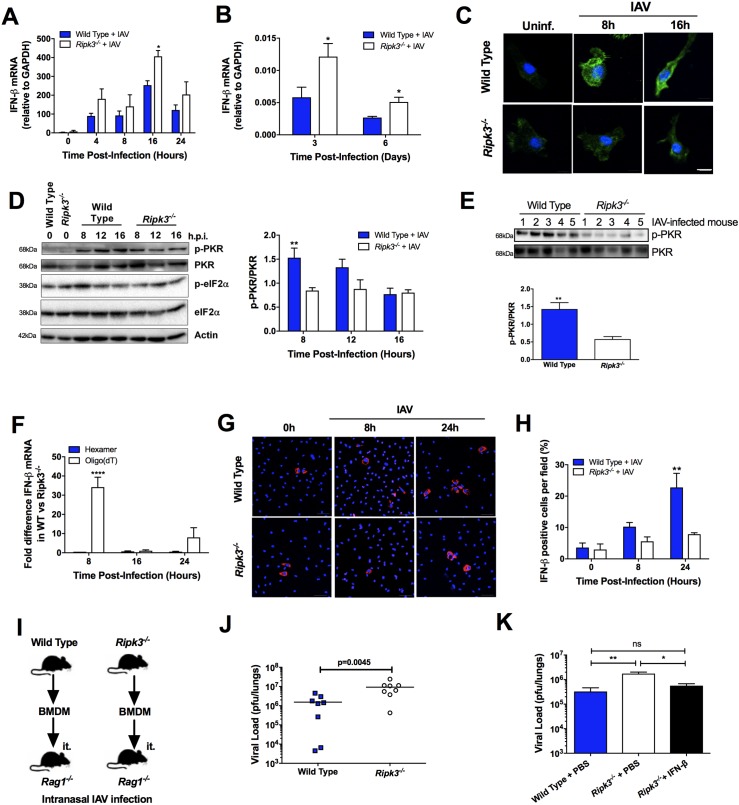
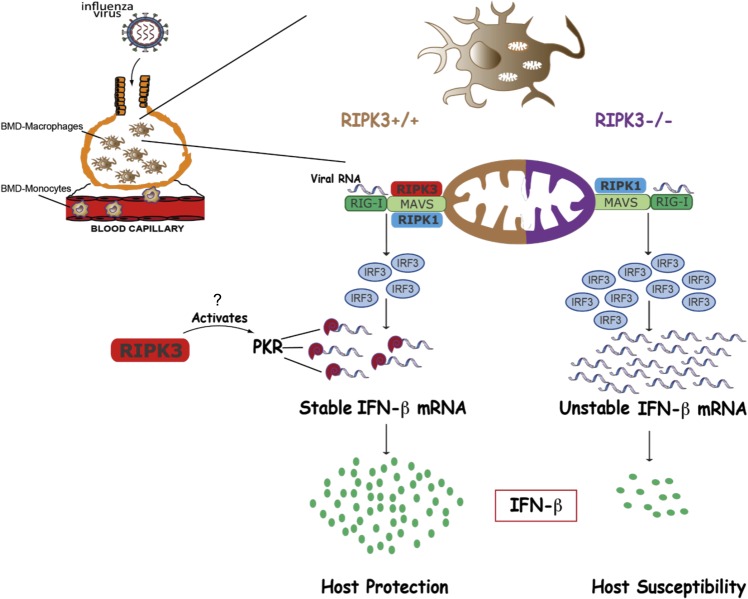
The type I interferon pathway plays a critical role in both host defense and tolerance against viral infection and thus requires refined regulatory mechanisms. RIPK3-mediated necroptosis has been shown to be involved in anti-viral immunity. However, the exact role of RIPK3 in immunity to Influenza A Virus (IAV) is poorly understood. In line with others, we, herein, show that Ripk3-/- mice are highly susceptible to IAV infection, exhibiting elevated pulmonary viral load and heightened morbidity and mortality. Unexpectedly, this susceptibility was linked to an inability of RIKP3-deficient macrophages (Mφ) to produce type I IFN in the lungs of infected mice. In Mφ infected with IAV in vitro, we found that RIPK3 regulates type I IFN both transcriptionally, by interacting with MAVS and limiting RIPK1 interaction with MAVS, and post-transcriptionally, by activating protein kinase R (PKR)-a critical regulator of IFN-β mRNA stability. Collectively, our findings indicate a novel role for RIPK3 in regulating Mφ-mediated type I IFN anti-viral immunity, independent of its conventional role in necroptosis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, et al. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27(2):240–52. doi: 10.1016/j.immuni.2007.07.013 - DOI - PubMed
-
- Wang J, Nikrad MP, Travanty EA, Zhou B, Phang T, Gao B, et al. Innate immune response of human alveolar macrophages during influenza A infection. PLoS One. 2012;7(3):e29879 Epub 2012 Mar 2. doi: 10.1371/journal.pone.0029879 - DOI - PMC - PubMed
-
- Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med. 2015;212(5):699–714. Epub 2015 Apr 20. doi: 10.1084/jem.20140825 - DOI - PMC - PubMed
-
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347(6227):aaa2630 doi: 10.1126/science.aaa2630 - DOI - PubMed
-
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13(1):129–41. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
