

iPSCORE: A Resource of 222 iPSC Lines Enabling Functional Characterization of Genetic Variation across a Variety of Cell Types
- PMID: 28410642
- PMCID: PMC5390244
- DOI: 10.1016/j.stemcr.2017.03.012
iPSCORE: A Resource of 222 iPSC Lines Enabling Functional Characterization of Genetic Variation across a Variety of Cell Types
Abstract
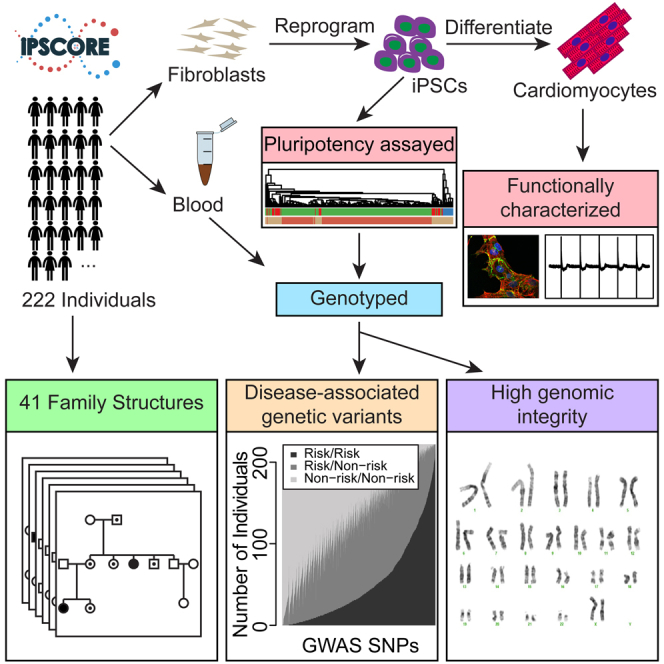
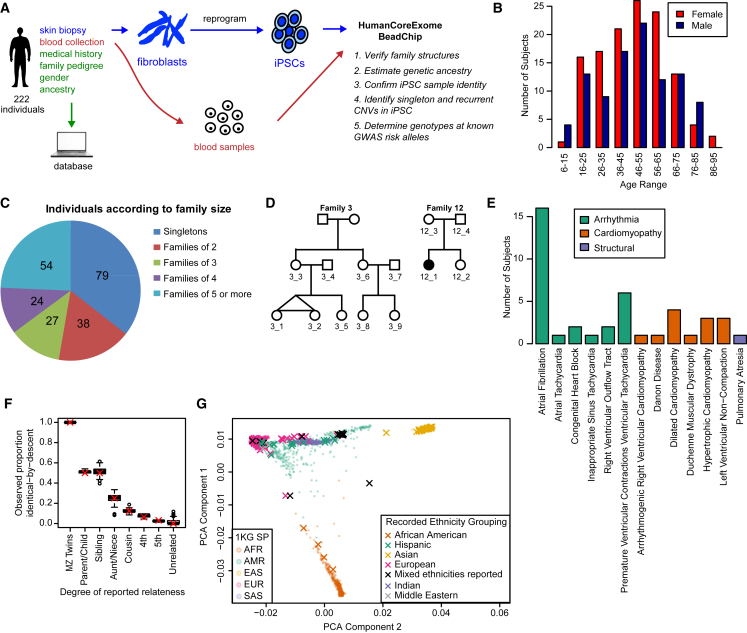
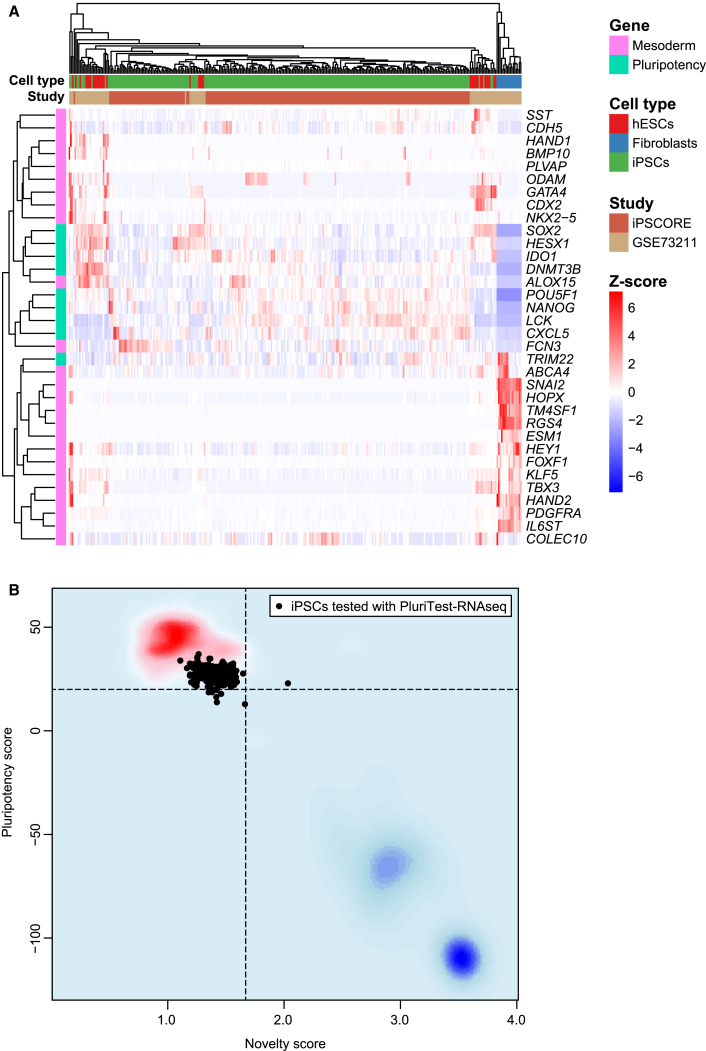
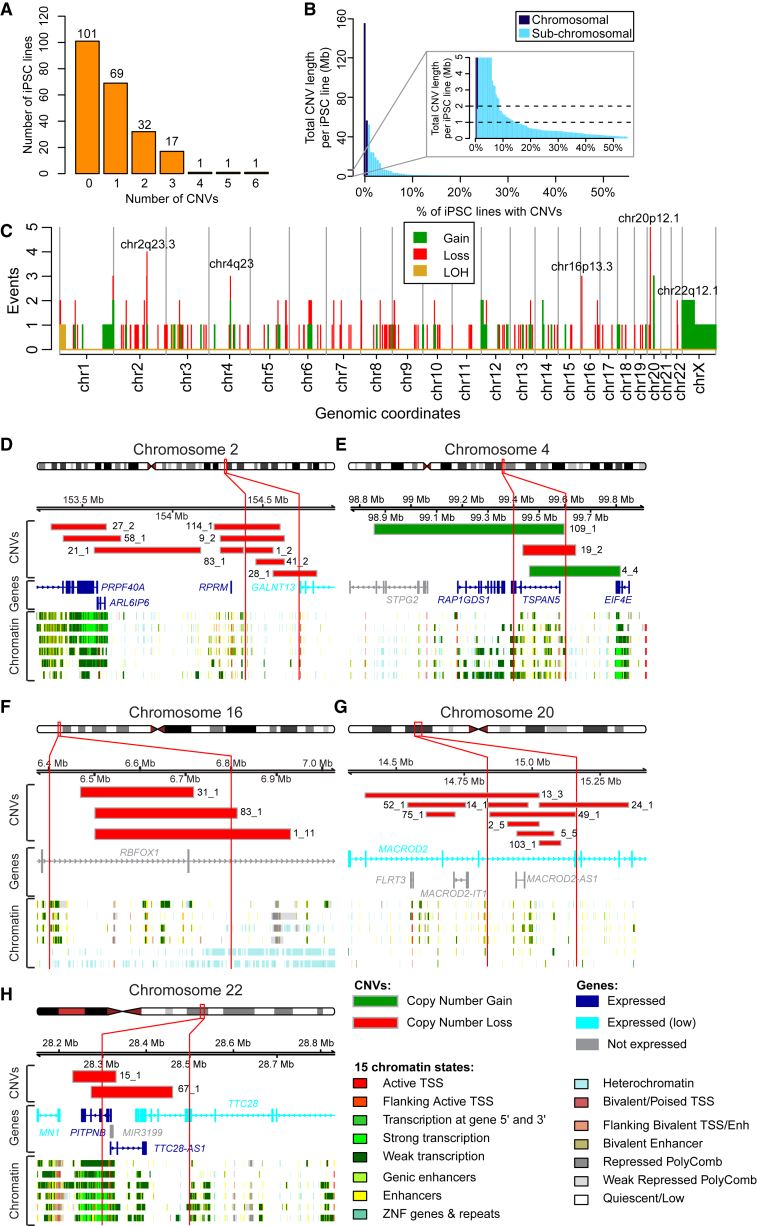
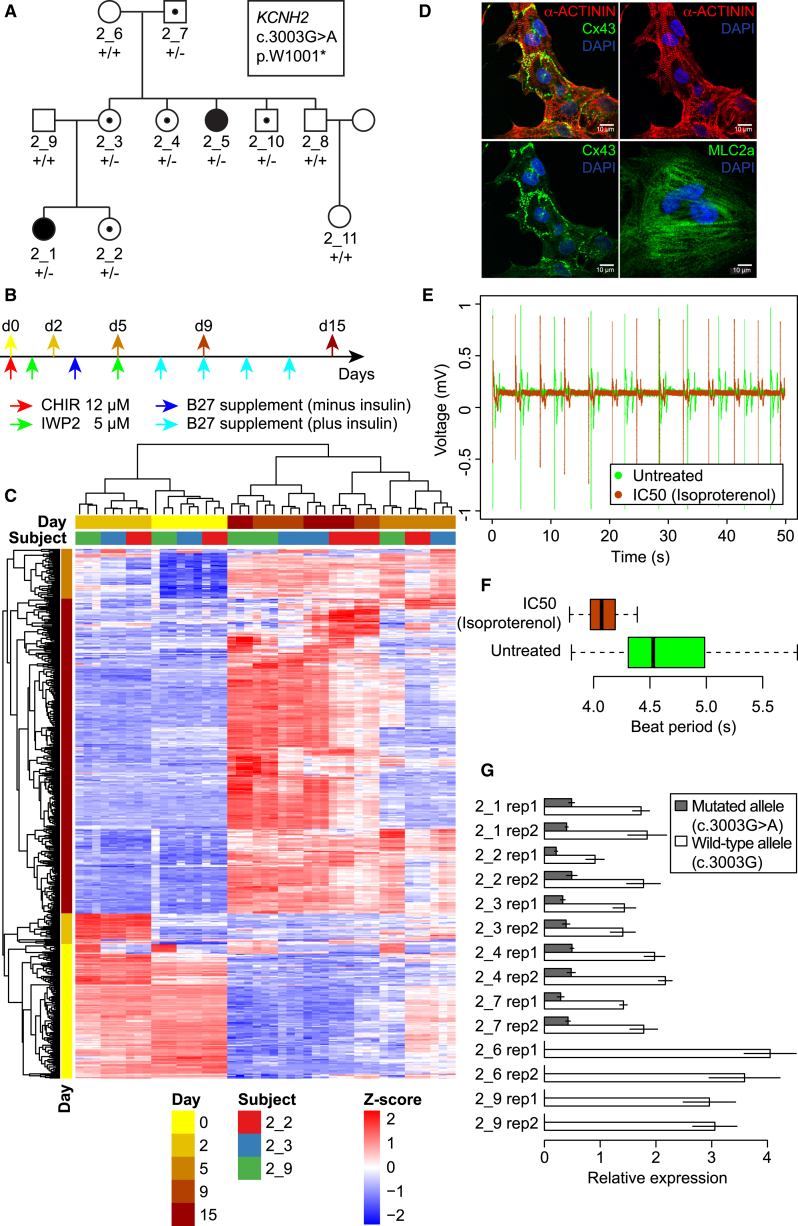
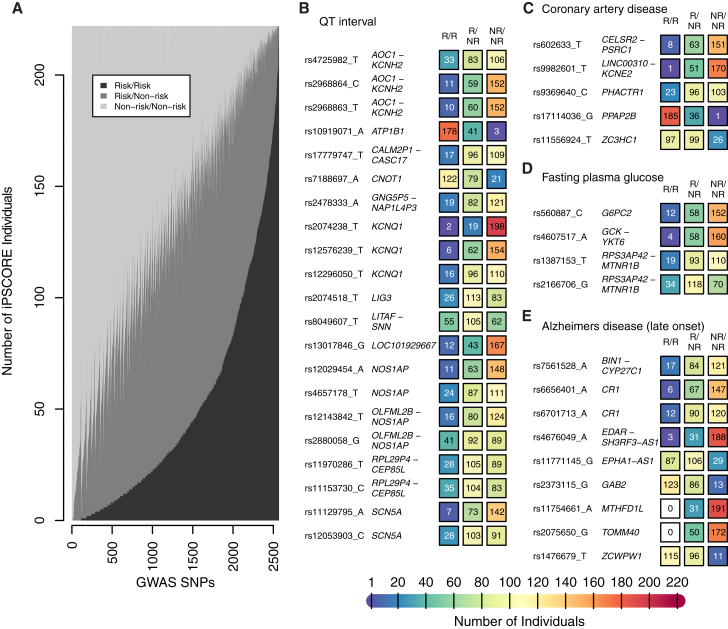
Large-scale collections of induced pluripotent stem cells (iPSCs) could serve as powerful model systems for examining how genetic variation affects biology and disease. Here we describe the iPSCORE resource: a collection of systematically derived and characterized iPSC lines from 222 ethnically diverse individuals that allows for both familial and association-based genetic studies. iPSCORE lines are pluripotent with high genomic integrity (no or low numbers of somatic copy-number variants) as determined using high-throughput RNA-sequencing and genotyping arrays, respectively. Using iPSCs from a family of individuals, we show that iPSC-derived cardiomyocytes demonstrate gene expression patterns that cluster by genetic background, and can be used to examine variants associated with physiological and disease phenotypes. The iPSCORE collection contains representative individuals for risk and non-risk alleles for 95% of SNPs associated with human phenotypes through genome-wide association studies. Our study demonstrates the utility of iPSCORE for examining how genetic variants influence molecular and physiological traits in iPSCs and derived cell lines.
Keywords: GWAS; KCNH2; LQT2; NHLBI Next Gen; cardiac disease; iPSC; iPSC-derived cardiomyocytes; iPSCORE; molecular traits; physiological traits.
Published by Elsevier Inc.
Figures
References
-
- Avior Y., Sagi I., Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016;17:170–182. - PubMed
MeSH terms
Grants and funding
- U01 DK105541/DK/NIDDK NIH HHS/United States
- T32 GM008806/GM/NIGMS NIH HHS/United States
- R01 EY021237/EY/NEI NIH HHS/United States
- UM1 HL128773/HL/NHLBI NIH HHS/United States
- UL1 TR001442/TR/NCATS NIH HHS/United States
- DP3 DK112155/DK/NIDDK NIH HHS/United States
- R41 HG008118/HG/NHGRI NIH HHS/United States
- U01 HL107442/HL/NHLBI NIH HHS/United States
- T15 LM011271/LM/NLM NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- T32 GM008666/GM/NIGMS NIH HHS/United States
- R44 HG008118/HG/NHGRI NIH HHS/United States
- UL1 TR000100/TR/NCATS NIH HHS/United States
- P30 CA014195/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
