

Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases
- PMID: 28411118
- PMCID: PMC5522763
- DOI: 10.1016/j.nbd.2017.04.010
Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases
Abstract
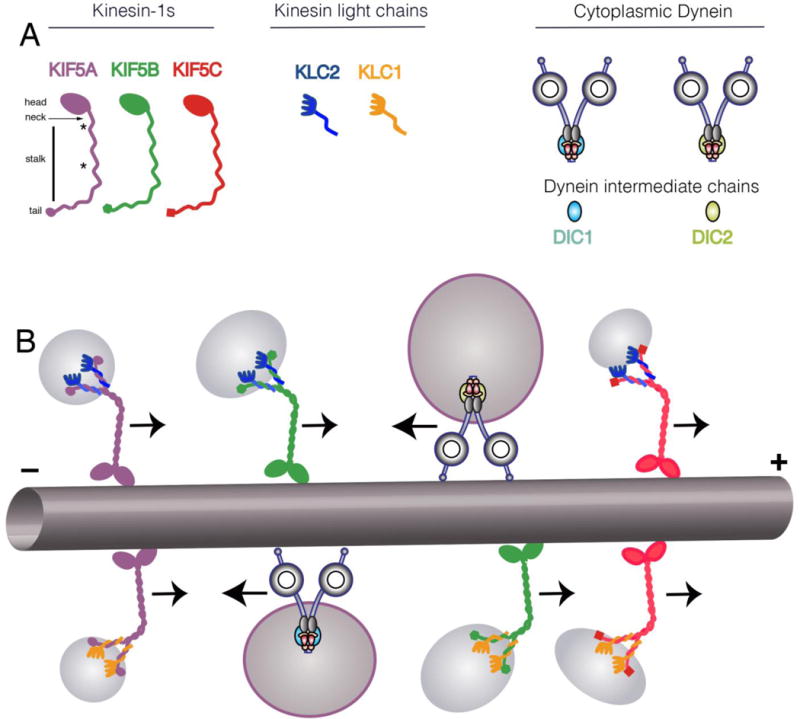
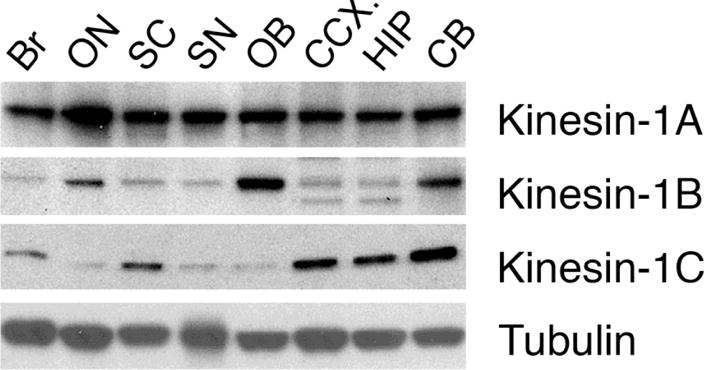
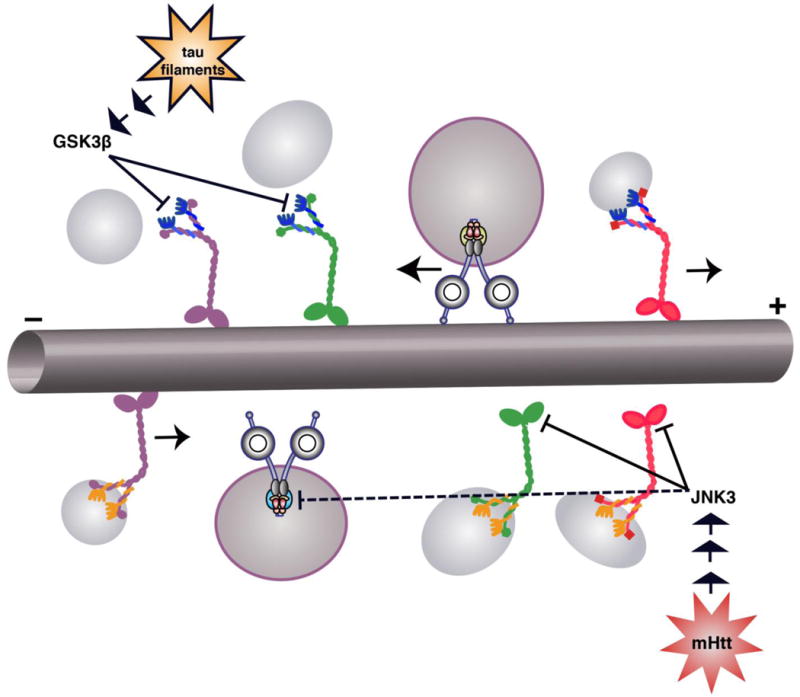
Neurons affected in a wide variety of unrelated adult-onset neurodegenerative diseases (AONDs) typically exhibit a "dying back" pattern of degeneration, which is characterized by early deficits in synaptic function and neuritic pathology long before neuronal cell death. Consistent with this observation, multiple unrelated AONDs including Alzheimer's disease, Parkinson's disease, Huntington's disease, and several motor neuron diseases feature early alterations in kinase-based signaling pathways associated with deficits in axonal transport (AT), a complex cellular process involving multiple intracellular trafficking events powered by microtubule-based motor proteins. These pathogenic events have important therapeutic implications, suggesting that a focus on preservation of neuronal connections may be more effective to treat AONDs than addressing neuronal cell death. While the molecular mechanisms underlying AT abnormalities in AONDs are still being analyzed, evidence has accumulated linking those to a well-established pathological hallmark of multiple AONDs: altered patterns of neuronal protein phosphorylation. Here, we present a short overview on the biochemical heterogeneity of major motor proteins for AT, their regulation by protein kinases, and evidence revealing cell type-specific AT specializations. When considered together, these findings may help explain how independent pathogenic pathways can affect AT differentially in the context of each AOND.
Keywords: Alzheimer's disease; Amyotrophic lateral sclerosis; Axonal transport; Dynen; Huntington's disease; Kinases; Kinesin; Neurodegeneration; Parkinson's disease; Signaling.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Ackerley S, et al. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2004;26:354–64. - PubMed
-
- Adalbert R, Coleman MP. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2013;39:90–108. - PubMed
-
- Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:167–79. - PubMed
-
- Bellucci A, et al. Review: Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42:77–94. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
