

Posttranslationally modified progesterone receptors direct ligand-specific expression of breast cancer stem cell-associated gene programs
- PMID: 28412963
- PMCID: PMC5392969
- DOI: 10.1186/s13045-017-0462-7
Posttranslationally modified progesterone receptors direct ligand-specific expression of breast cancer stem cell-associated gene programs
Abstract
Background: Estrogen and progesterone are potent breast mitogens. In addition to steroid hormones, multiple signaling pathways input to estrogen receptor (ER) and progesterone receptor (PR) actions via posttranslational events. Protein kinases commonly activated in breast cancers phosphorylate steroid hormone receptors (SRs) and profoundly impact their activities.
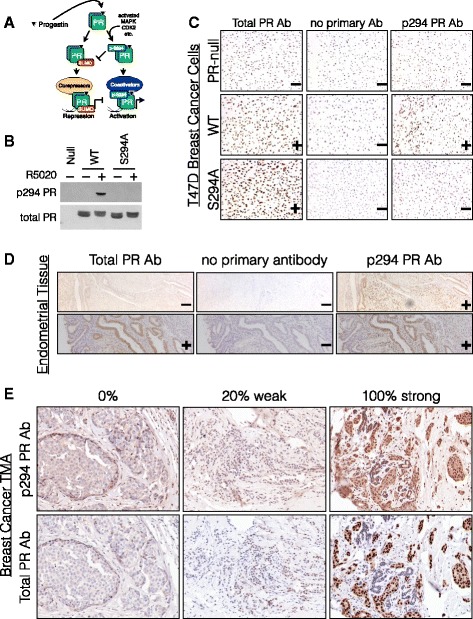
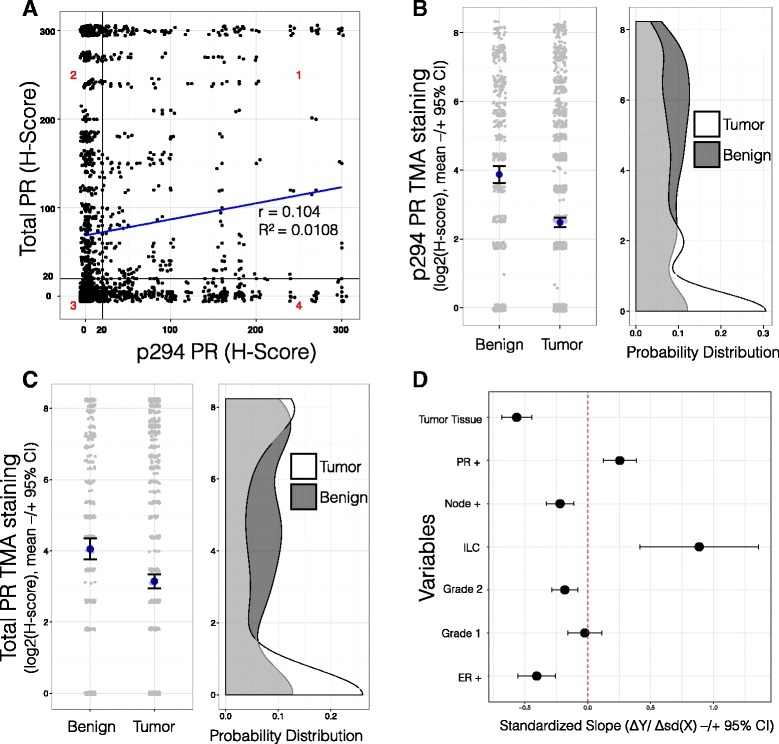
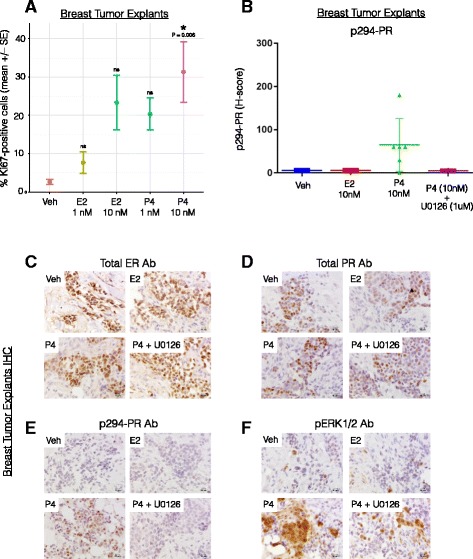
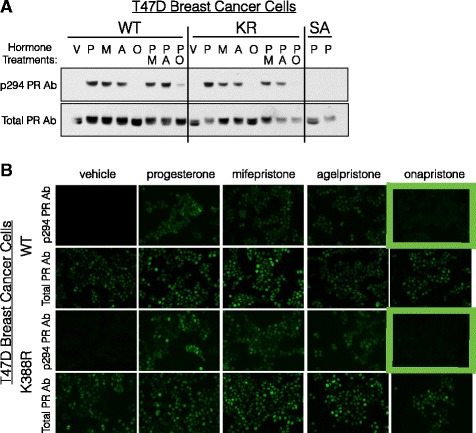
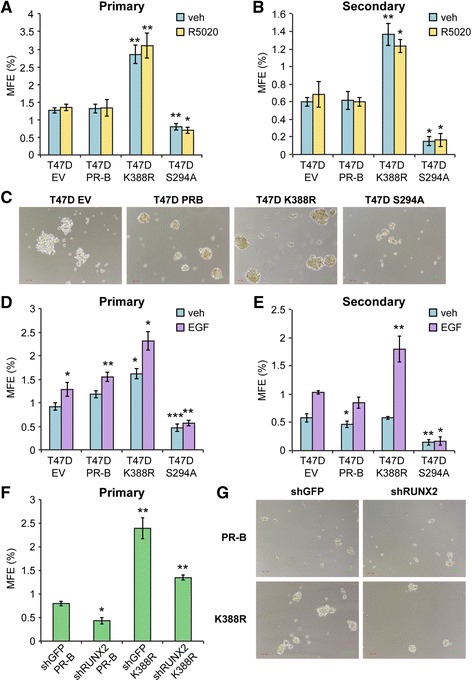
Methods: To better understand the role of modified PRs in breast cancer, we measured total and phospho-Ser294 PRs in 209 human breast tumors represented on 2754 individual tissue spots within a tissue microarray and assayed the regulation of this site in human tumor explants cultured ex vivo. To complement this analysis, we assayed PR target gene regulation in T47D luminal breast cancer models following treatment with progestin (promegestone; R5020) and antiprogestins (mifepristone, onapristone, or aglepristone) in conditions under which the receptor is regulated by Lys388 SUMOylation (K388 intact) or is SUMO-deficient (via K388R mutation to mimic persistent Ser294 phosphorylation). Selected phospho-PR-driven target genes were validated by qRT-PCR and following RUNX2 shRNA knockdown in breast cancer cell lines. Primary and secondary mammosphere assays were performed to implicate phospho-Ser294 PRs, epidermal growth factor signaling, and RUNX2 in breast cancer stem cell biology.
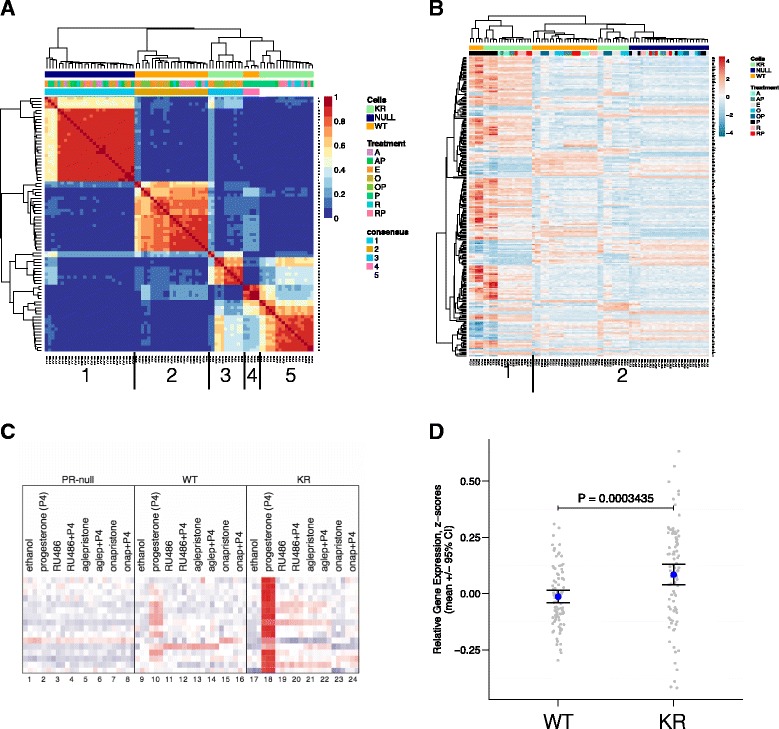
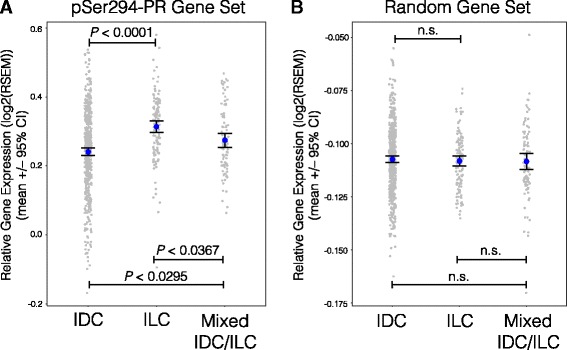
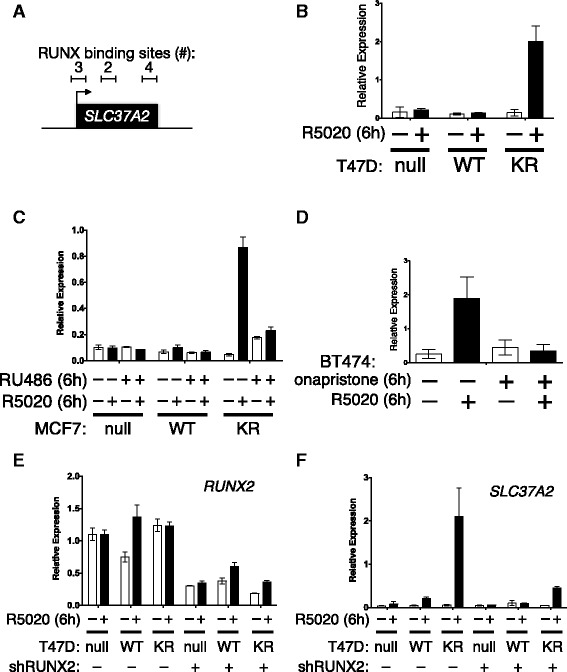
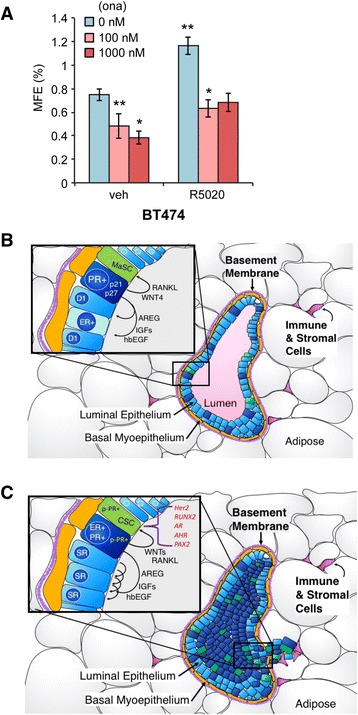
Results: Phospho-Ser294 PR species were abundant in a majority (54%) of luminal breast tumors, and PR promoter selectivity was exquisitely sensitive to posttranslational modifications. Phospho-PR expression and target gene programs were significantly associated with invasive lobular carcinoma (ILC). Consistent with our finding that activated phospho-PRs undergo rapid ligand-dependent turnover, unique phospho-PR gene signatures were most prevalent in breast tumors clinically designated as PR-low to PR-null (luminal B) and included gene sets associated with cancer stem cell biology (HER2, PAX2, AHR, AR, RUNX). Validation studies demonstrated a requirement for RUNX2 in the regulation of selected phospho-PR target genes (SLC37A2). In vitro mammosphere formation assays support a role for phospho-Ser294-PRs via growth factor (EGF) signaling as well as RUNX2 as potent drivers of breast cancer stem cell fate.
Conclusions: We conclude that PR Ser294 phosphorylation is a common event in breast cancer progression that is required to maintain breast cancer stem cell fate, in part via cooperation with growth factor-initiated signaling pathways and key phospho-PR target genes including SLC37A2 and RUNX2. Clinical measurement of phosphorylated PRs should be considered a useful marker of breast tumor stem cell potential. Alternatively, unique phospho-PR target gene sets may provide useful tools with which to identify patients likely to respond to selective PR modulators that block PR Ser294 phosphorylation as part of rational combination (i.e., with antiestrogens) endocrine therapies designed to durably block breast cancer recurrence.
Keywords: Antiprogestin; Breast cancer; Cancer stem cells; ERK/MAP kinase (MAPK); Estrogen receptor (ER); Onapristone; Phosphorylation; Progesterone receptor (PR); RUNX2; SUMOylation.
Figures
Similar articles
-
Phosphorylated and sumoylation-deficient progesterone receptors drive proliferative gene signatures during breast cancer progression.Breast Cancer Res. 2012 Jun 14;14(3):R95. doi: 10.1186/bcr3211. Breast Cancer Res. 2012. PMID: 22697792 Free PMC article.
-
Phosphorylation-dependent antagonism of sumoylation derepresses progesterone receptor action in breast cancer cells.Mol Endocrinol. 2007 Dec;21(12):2890-906. doi: 10.1210/me.2007-0248. Epub 2007 Aug 23. Mol Endocrinol. 2007. PMID: 17717077
-
Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth.Steroids. 2007 Feb;72(2):188-201. doi: 10.1016/j.steroids.2006.11.009. Epub 2006 Dec 14. Steroids. 2007. PMID: 17173941 Free PMC article.
-
Tracking progesterone receptor-mediated actions in breast cancer.Pharmacol Ther. 2014 Apr;142(1):114-25. doi: 10.1016/j.pharmthera.2013.11.010. Epub 2013 Nov 26. Pharmacol Ther. 2014. PMID: 24291072 Free PMC article. Review.
-
90 YEARS OF PROGESTERONE: Steroid receptors as MAPK signaling sensors in breast cancer: let the fates decide.J Mol Endocrinol. 2020 Jul;65(1):T35-T48. doi: 10.1530/JME-19-0274. J Mol Endocrinol. 2020. PMID: 32209723 Free PMC article. Review.
Cited by
-
Involvement of the Estrogen and Progesterone Axis in Cancer Stemness: Elucidating Molecular Mechanisms and Clinical Significance.Front Oncol. 2020 Sep 4;10:1657. doi: 10.3389/fonc.2020.01657. eCollection 2020. Front Oncol. 2020. PMID: 33014829 Free PMC article. Review.
-
Onapristone Extended Release: Safety Evaluation from Phase I-II Studies with an Emphasis on Hepatotoxicity.Drug Saf. 2020 Oct;43(10):1045-1055. doi: 10.1007/s40264-020-00964-x. Drug Saf. 2020. PMID: 32594454 Free PMC article.
-
Impaired redox regulation of estrogen metabolizing proteins is important determinant of human breast cancers.Cancer Cell Int. 2019 May 15;19:111. doi: 10.1186/s12935-019-0826-x. eCollection 2019. Cancer Cell Int. 2019. PMID: 31114446 Free PMC article. Review.
-
O-GlcNAc-Dependent Regulation of Progesterone Receptor Function in Breast Cancer.Horm Cancer. 2018 Feb;9(1):12-21. doi: 10.1007/s12672-017-0310-9. Epub 2017 Sep 19. Horm Cancer. 2018. PMID: 28929346 Free PMC article.
-
Microenvironmental Determinants of Breast Cancer Metastasis: Focus on the Crucial Interplay Between Estrogen and Insulin/Insulin-Like Growth Factor Signaling.Front Cell Dev Biol. 2020 Dec 8;8:608412. doi: 10.3389/fcell.2020.608412. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33364239 Free PMC article. Review.
References
-
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. - DOI - PMC - PubMed
-
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. - PMC - PubMed
-
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Caldas C, Aparicio S, Curtis C, Shah SP, Caldas C, Aparicio S, Brenton JD, Ellis I, Huntsman D, Pinder S, Purushotham A, Murphy L, Caldas C, Aparicio S et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
