

Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability
- PMID: 28414775
- PMCID: PMC5393878
- DOI: 10.1371/journal.pone.0175962
Xp11.22 deletions encompassing CENPVL1, CENPVL2, MAGED1 and GSPT2 as a cause of syndromic X-linked intellectual disability
Abstract
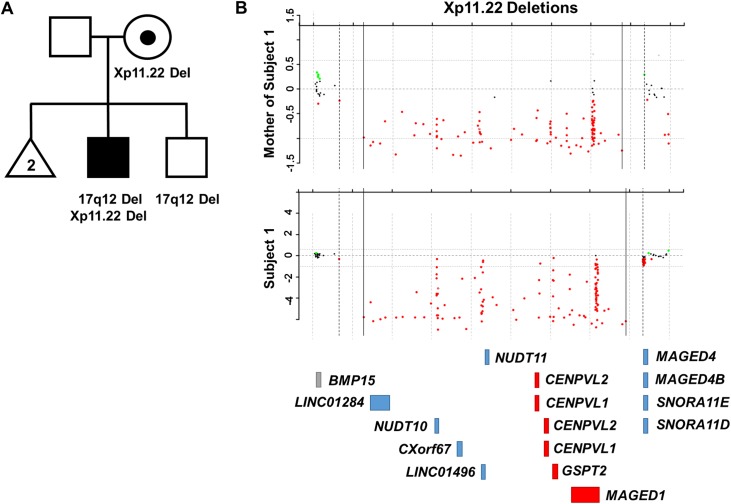
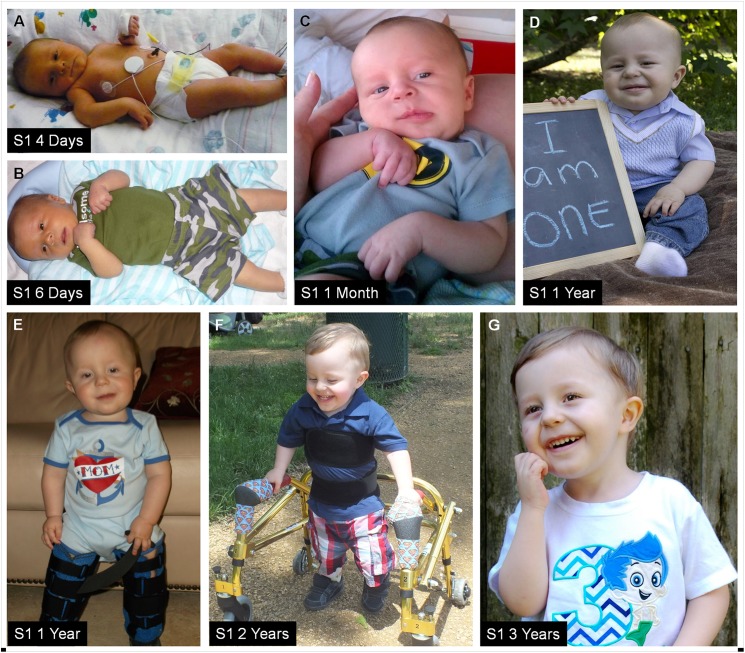
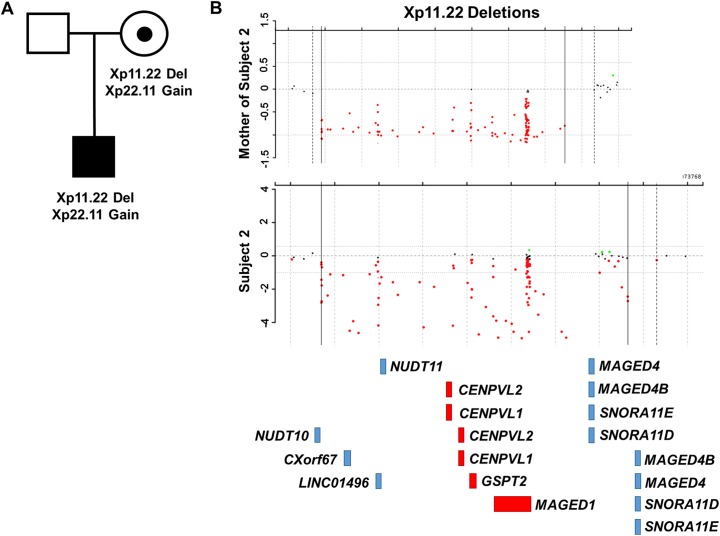
By searching a clinical database of over 60,000 individuals referred for array-based CNV analyses and online resources, we identified four males from three families with intellectual disability, developmental delay, hypotonia, joint hypermobility and relative macrocephaly who carried small, overlapping deletions of Xp11.22. The maximum region of overlap between their deletions spanned ~430 kb and included two pseudogenes, CENPVL1 and CENPVL2, whose functions are not known, and two protein coding genes-the G1 to S phase transition 2 gene (GSPT2) and the MAGE family member D1 gene (MAGED1). Deletions of this ~430 kb region have not been previously implicated in human disease. Duplications of GSPT2 have been documented in individuals with intellectual disability, but the phenotypic consequences of a loss of GSPT2 function have not been elucidated in humans or mouse models. Changes in MAGED1 have not been associated with intellectual disability in humans, but loss of MAGED1 function is associated with neurocognitive and neurobehavioral phenotypes in mice. In all cases, the Xp11.22 deletion was inherited from an unaffected mother. Studies performed on DNA from one of these mothers did not show evidence of skewed X-inactivation. These results suggest that deletions of an ~430 kb region on chromosome Xp11.22 that encompass CENPVL1, CENPVL2, GSPT2 and MAGED1 cause a distinct X-linked syndrome characterized by intellectual disability, developmental delay, hypotonia, joint hypermobility and relative macrocephaly. Loss of GSPT2 and/or MAGED1 function may contribute to the intellectual disability and developmental delay seen in males with these deletions.
Conflict of interest statement
Figures

Similar articles
-
Further Clinical and Molecular Delineation of Xp11.22 Deletion Syndrome: A Case Report.Oman Med J. 2019 Sep;34(5):460-463. doi: 10.5001/omj.2019.83. Oman Med J. 2019. PMID: 31555424 Free PMC article.
-
Chromosome Xq28 duplication encompassing MECP2: Clinical and molecular analysis of 16 new patients from 10 families in China.Eur J Med Genet. 2016 Jun;59(6-7):347-53. doi: 10.1016/j.ejmg.2016.05.004. Epub 2016 May 11. Eur J Med Genet. 2016. PMID: 27180140
-
Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions.J Med Genet. 2011 Dec;48(12):840-50. doi: 10.1136/jmedgenet-2011-100125. Epub 2011 Oct 8. J Med Genet. 2011. PMID: 21984752
-
Duplication Xp11.22-p14 in females: does X-inactivation help in assessing their significance?Am J Med Genet A. 2015 Mar;167A(3):553-62. doi: 10.1002/ajmg.a.36897. Am J Med Genet A. 2015. PMID: 25691408 Review.
-
Phenotype-genotype correlations in 17 new patients with an Xp11.23p11.22 microduplication and review of the literature.Am J Med Genet A. 2015 Jan;167A(1):111-22. doi: 10.1002/ajmg.a.36807. Epub 2014 Nov 25. Am J Med Genet A. 2015. PMID: 25425167 Review.
Cited by
-
Characterization of large-scale genomic differences in the first complete human genome.Genome Biol. 2023 Jul 4;24(1):157. doi: 10.1186/s13059-023-02995-w. Genome Biol. 2023. PMID: 37403156 Free PMC article.
-
Deletion of Maged1 in mice abolishes locomotor and reinforcing effects of cocaine.EMBO Rep. 2018 Sep;19(9):e45089. doi: 10.15252/embr.201745089. Epub 2018 Jul 12. EMBO Rep. 2018. PMID: 30002119 Free PMC article.
-
Identification of a Locus on the X Chromosome Linked to Familial Membranous Nephropathy.Kidney Int Rep. 2021 Mar 3;6(6):1669-1676. doi: 10.1016/j.ekir.2021.02.025. eCollection 2021 Jun. Kidney Int Rep. 2021. PMID: 34169208 Free PMC article.
-
Identification of novel candidate disease genes from de novo exonic copy number variants.Genome Med. 2017 Sep 21;9(1):83. doi: 10.1186/s13073-017-0472-7. Genome Med. 2017. PMID: 28934986 Free PMC article.
-
Genetic Analysis of the X Chromosome Associates Loci with Progression of Parkinson's Disease.Mov Disord. 2025 Jun 3:10.1002/mds.30252. doi: 10.1002/mds.30252. Online ahead of print. Mov Disord. 2025. PMID: 40459076
References
-
- Qiao Y, Liu X, Harvard C, Hildebrand MJ, Rajcan-Separovic E, Holden JJ, et al. (2008) Autism-associated familial microdeletion of Xp11.22. Clin Genet 74: 134–144. doi: 10.1111/j.1399-0004.2008.01028.x - DOI - PubMed
-
- Giorda R, Bonaglia MC, Beri S, Fichera M, Novara F, Magini P, et al. (2009) Complex segmental duplications mediate a recurrent dup(X)(p11.22-p11.23) associated with mental retardation, speech delay, and EEG anomalies in males and females. Am J Hum Genet 85: 394–400. doi: 10.1016/j.ajhg.2009.08.001 - DOI - PMC - PubMed
-
- Edens AC, Lyons MJ, Duron RM, Dupont BR, Holden KR (2011) Autism in two females with duplications involving Xp11.22-p11.23. Dev Med Child Neurol 53: 463–466. doi: 10.1111/j.1469-8749.2010.03909.x - DOI - PubMed
-
- Chung BH, Drmic I, Marshall CR, Grafodatskaya D, Carter M, Fernandez BA, et al. (2011) Phenotypic spectrum associated with duplication of Xp11.22-p11.23 includes Autism Spectrum Disorder. Eur J Med Genet 54: e516–520. doi: 10.1016/j.ejmg.2011.05.008 - DOI - PubMed
-
- Froyen G, Belet S, Martinez F, Santos-Reboucas CB, Declercq M, Verbeeck J, et al. (2012) Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am J Hum Genet 91: 252–264. doi: 10.1016/j.ajhg.2012.06.010 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
