

Inhibitor SBFI26 suppresses the malignant progression of castration-resistant PC3-M cells by competitively binding to oncogenic FABP5
- PMID: 28415688
- PMCID: PMC5458187
- DOI: 10.18632/oncotarget.16055
Inhibitor SBFI26 suppresses the malignant progression of castration-resistant PC3-M cells by competitively binding to oncogenic FABP5
Erratum in
-
Correction: Inhibitor SBFI26 suppresses the malignant progression of castration-resistant PC3-M cells by competitively binding to oncogenic FABP5.Oncotarget. 2020 Aug 4;11(31):3025. doi: 10.18632/oncotarget.27690. eCollection 2020 Aug 4. Oncotarget. 2020. PMID: 32821347 Free PMC article.
Abstract
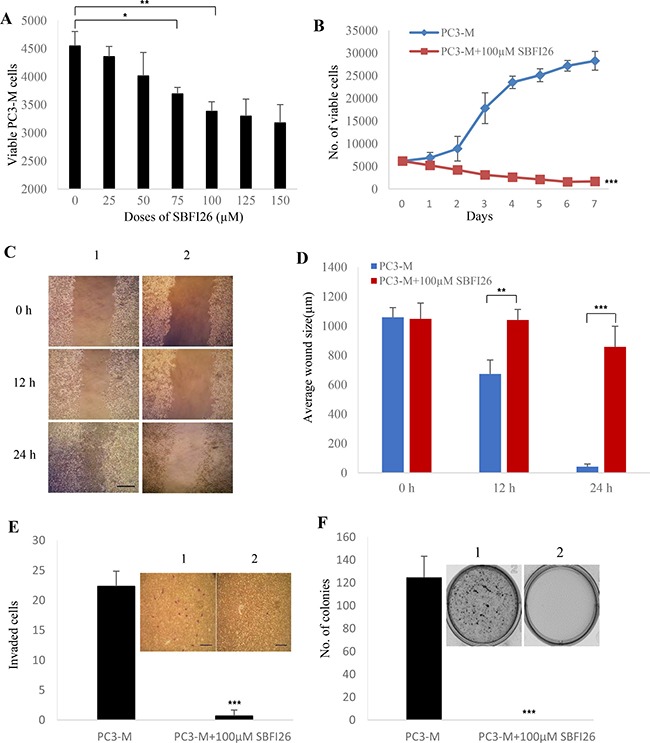
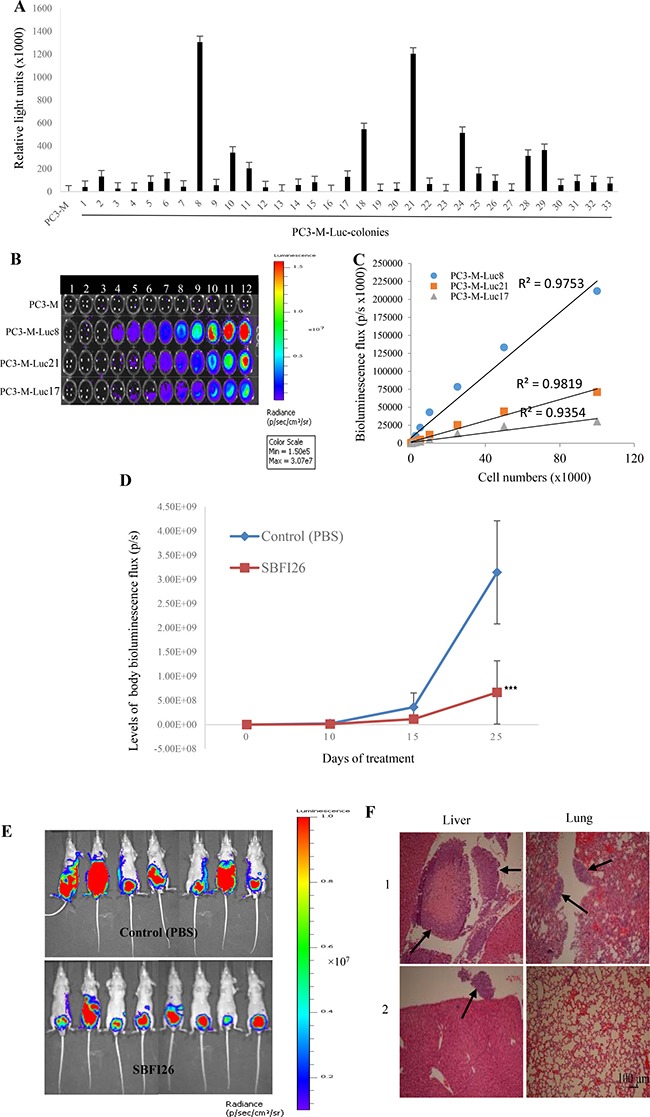
Castration resistant-prostate cancer is largely impervious to feather hormonal therapy and hence the outlook for patients is grim. Here we use an approach to attach the recently discovered Achilles heel. The experimental treatment established in this study is based on the recent discovery that it is the FABP5-PPARγ-VEGF signalling axis, rather than the androgen receptor pathway, played a dominant role in promoting the malignant progression of castration resistant prostate cancer cells. Treatments have been established in mice by suppressing the biological activity of FABP5 using a chemical inhibitor SBFI26. The inhibitor significantly suppressed the proliferation, migration, invasiveness and colony formation of PC3-M cells in vitro. It also produced a highly significant suppression of both the metastases and the primary tumours developed from cancer cells implanted orthotopically into the prostate glands of the mice. The inhibitor SBFI26 interferes with the FABP5-PPARγ- signalling pathway at the initial stage of the signal transduction by binding competitively to FABP5 to inhibit cellular fatty acid uptake. This avoids the fatty-acid stimulation of PPARγ and prevents it activating the down-stream regulated cancer-promoting genes. This entirely novel experimental approach to treating castration- resistant prostate cancer is completely different from current treatments that are based on androgen-blockade therapy.
Keywords: CRPC; FABP5; PPARγ; SBFI26; metastasis.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures




References
-
- Ritch CR, Wan RL, Stephens LB, Taxy JB, Huo D, Gong EM, Zagaja GP, Brendler CB. Dietary fatty acids correlate with prostate cancer biopsy grade and volume in Jamaican men. J Urology. 2007;177:97–101. - PubMed
-
- Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: Current status and future prospects. Prostate. 2004;61:332–353. - PubMed
-
- Shah S, Carriveau WJ, Li J, Campbell SL, Kopinski PK, Lim HW, Daurio N, Trefely S, Won KJ, Wallace DC, Koumenis C, Mancuso A, Wellen KE. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget. 2016;7:43713–43730. doi: 10.18632/oncotarget.9666. - DOI - PMC - PubMed
-
- Wen S, Niu Y, Lee SO, Yeh S, Shang Z, Gao H, Li Y, Chou F, Chang C. Targeting fatty acid synthase with ASC-J9 suppresses proliferation and invasion of prostate cancer cells. Mol carcinogenesis. 2016. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
