

Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism
- PMID: 28416471
- PMCID: PMC5501738
- DOI: 10.1158/2159-8290.CD-16-0441
Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism
Abstract
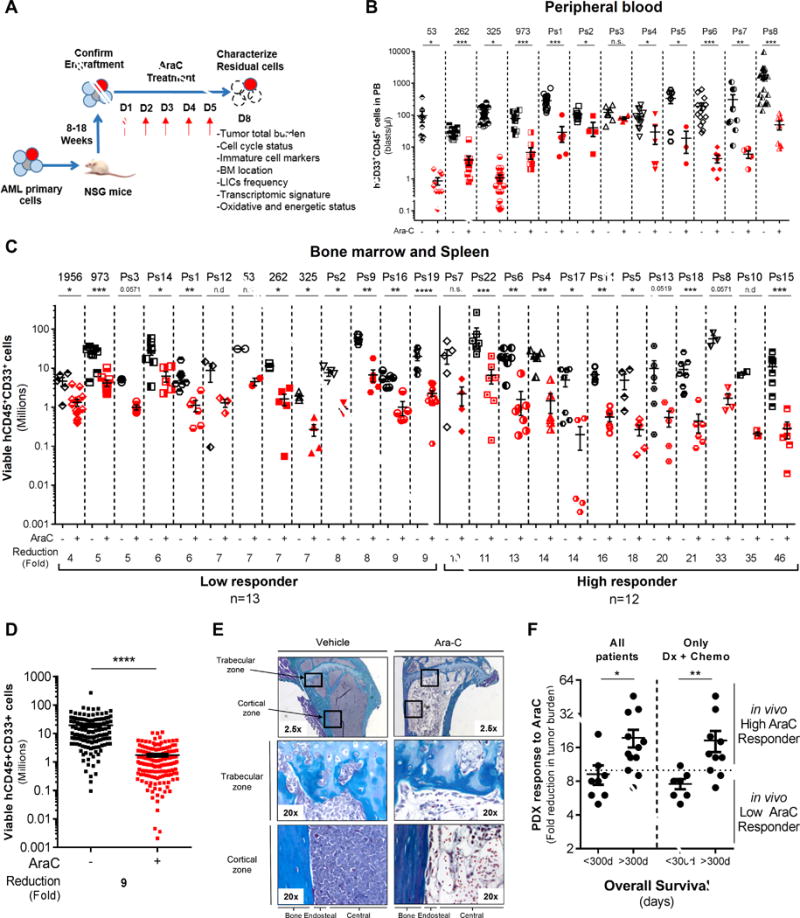
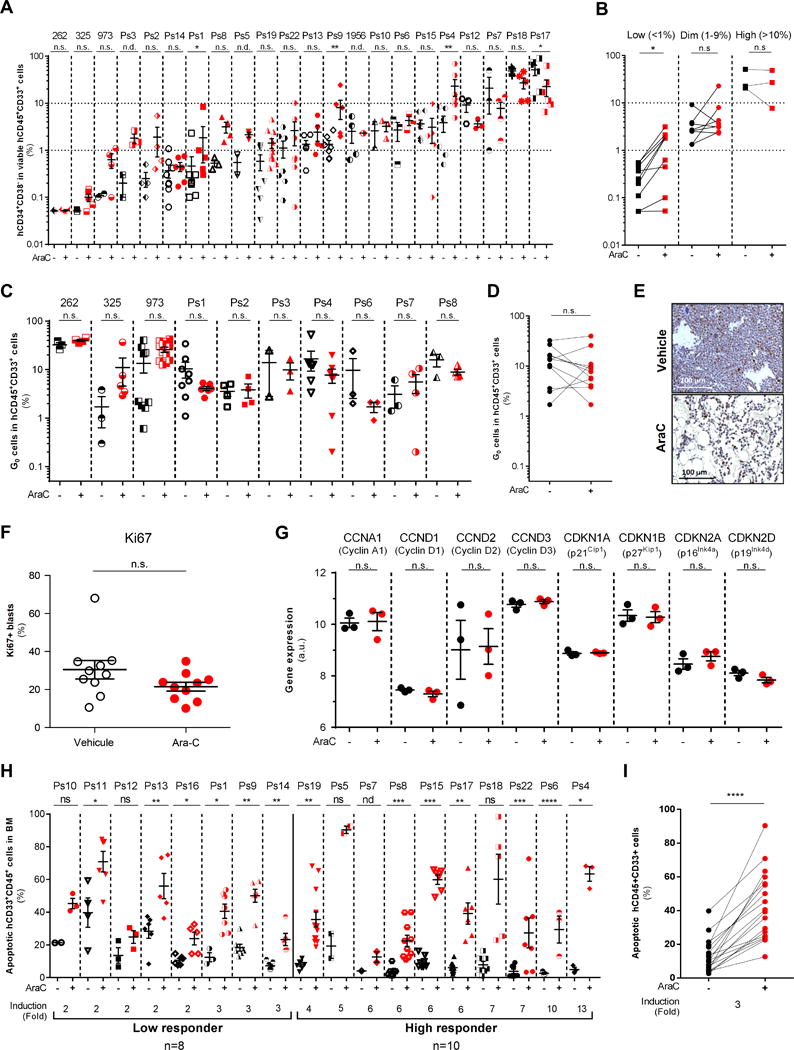
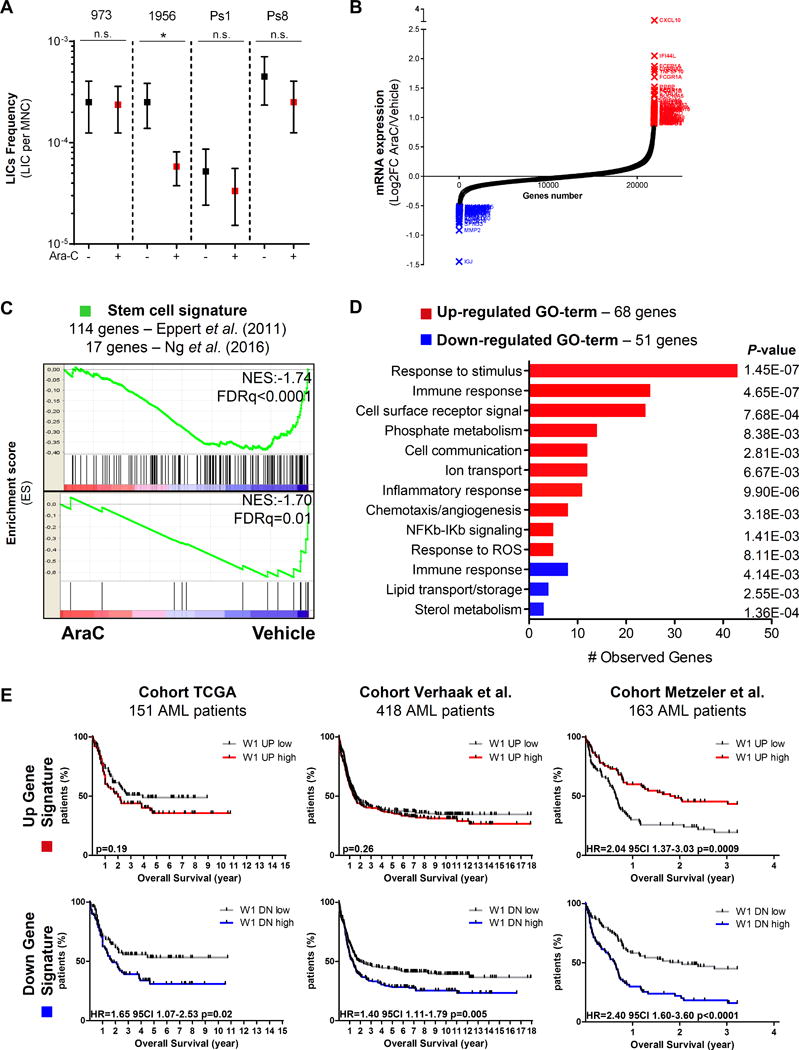
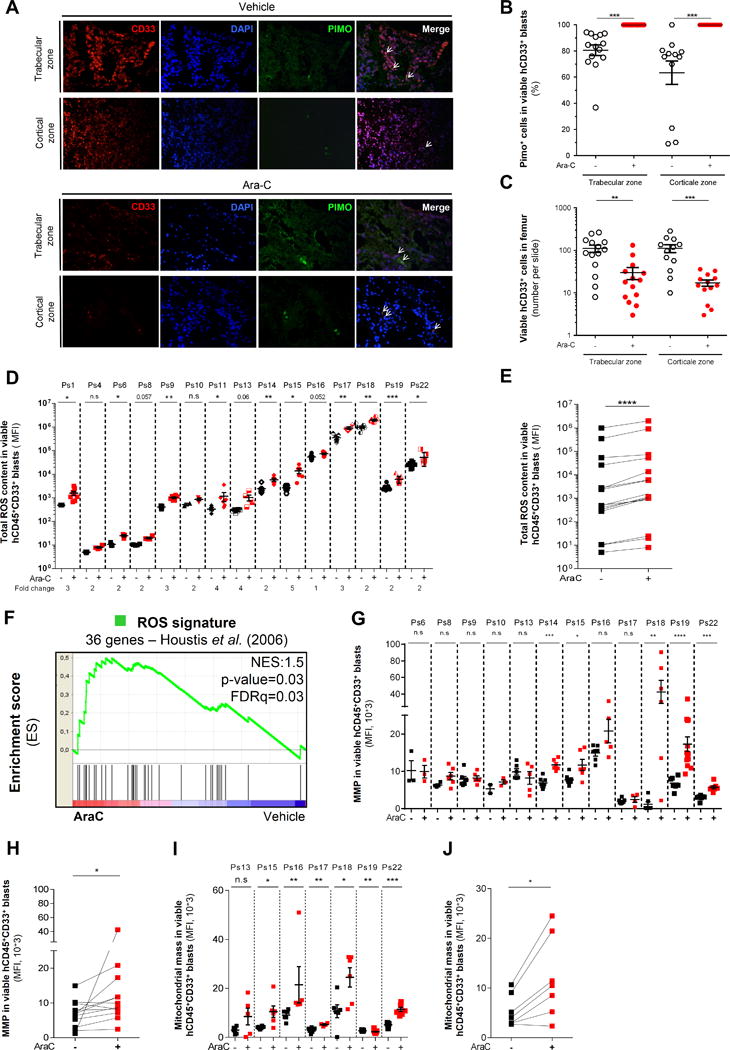
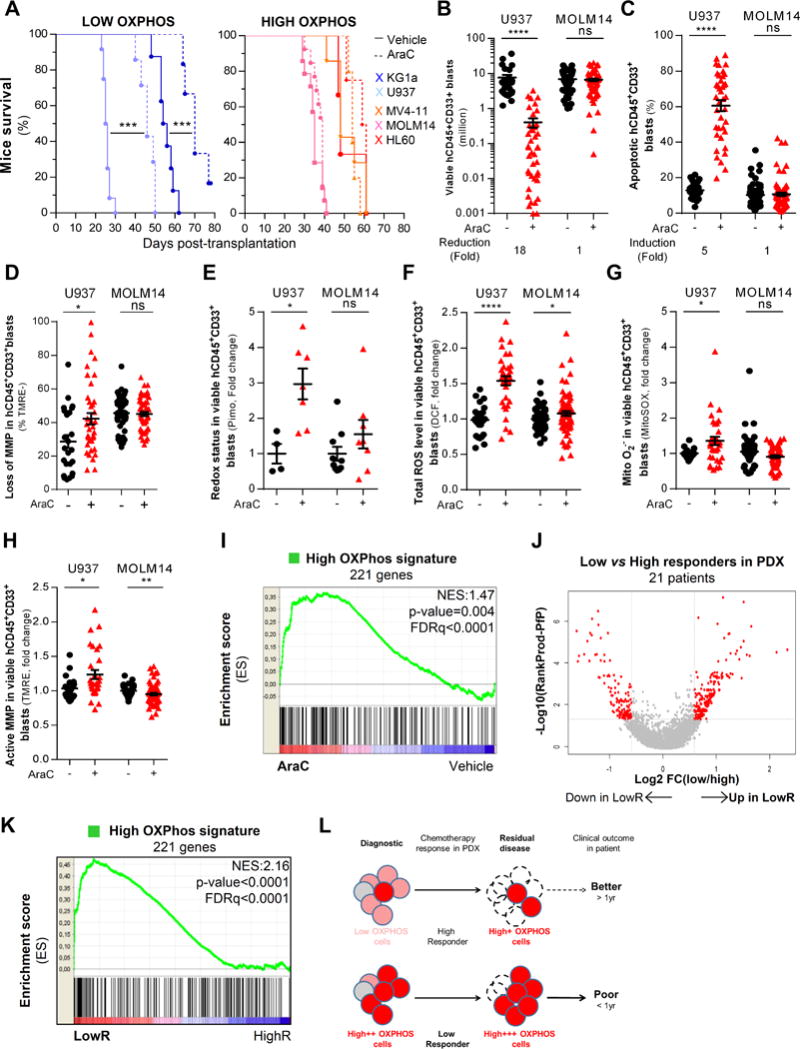
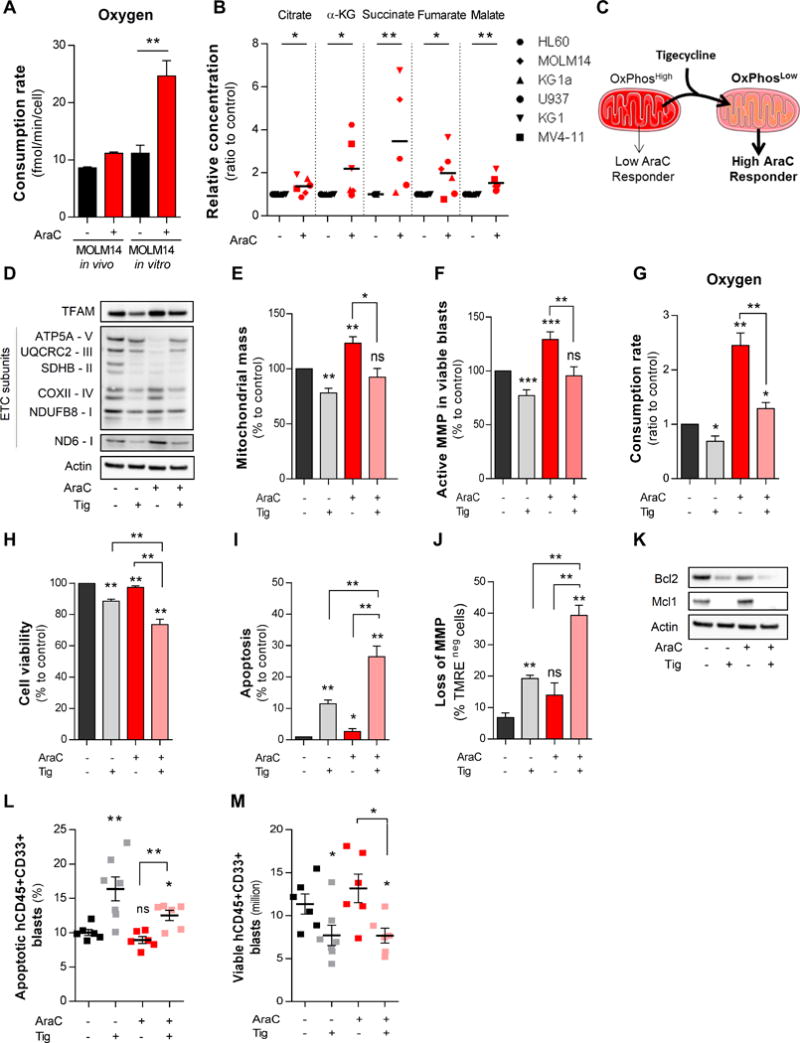
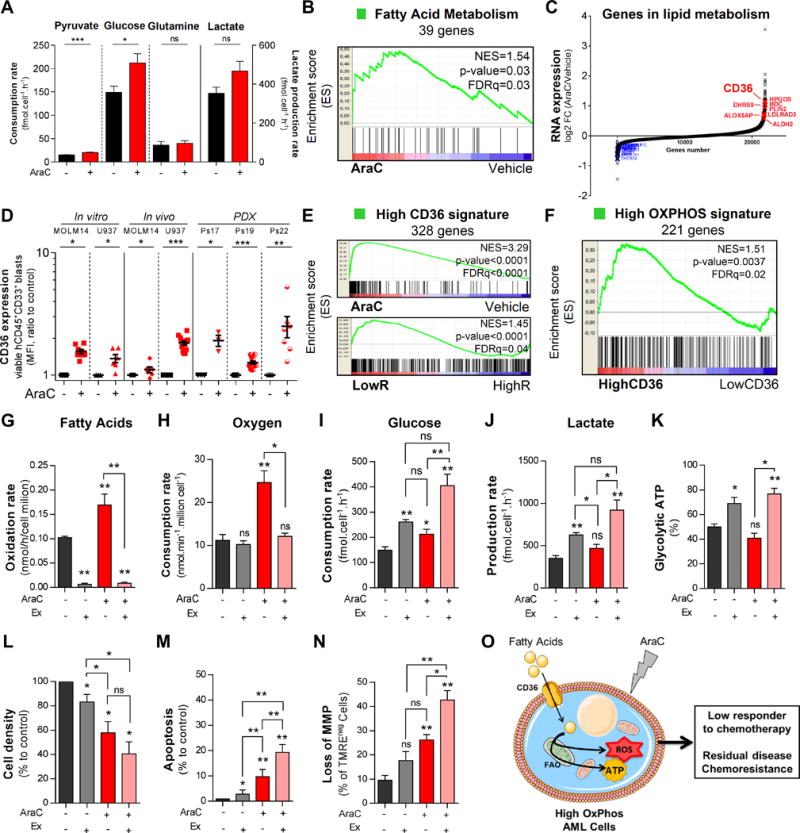
Chemotherapy-resistant human acute myeloid leukemia (AML) cells are thought to be enriched in quiescent immature leukemic stem cells (LSC). To validate this hypothesis in vivo, we developed a clinically relevant chemotherapeutic approach treating patient-derived xenografts (PDX) with cytarabine (AraC). AraC residual AML cells are enriched in neither immature, quiescent cells nor LSCs. Strikingly, AraC-resistant preexisting and persisting cells displayed high levels of reactive oxygen species, showed increased mitochondrial mass, and retained active polarized mitochondria, consistent with a high oxidative phosphorylation (OXPHOS) status. AraC residual cells exhibited increased fatty-acid oxidation, upregulated CD36 expression, and a high OXPHOS gene signature predictive for treatment response in PDX and patients with AML. High OXPHOS but not low OXPHOS human AML cell lines were chemoresistant in vivo. Targeting mitochondrial protein synthesis, electron transfer, or fatty-acid oxidation induced an energetic shift toward low OXPHOS and markedly enhanced antileukemic effects of AraC. Together, this study demonstrates that essential mitochondrial functions contribute to AraC resistance in AML and are a robust hallmark of AraC sensitivity and a promising therapeutic avenue to treat AML residual disease.Significance: AraC-resistant AML cells exhibit metabolic features and gene signatures consistent with a high OXPHOS status. In these cells, targeting mitochondrial metabolism through the CD36-FAO-OXPHOS axis induces an energetic shift toward low OXPHOS and strongly enhanced antileukemic effects of AraC, offering a promising avenue to design new therapeutic strategies and fight AraC resistance in AML. Cancer Discov; 7(7); 716-35. ©2017 AACR.See related commentary by Schimmer, p. 670This article is highlighted in the In This Issue feature, p. 653.
©2017 American Association for Cancer Research.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Novel Mitochondrial Mechanisms of Cytarabine Resistance in Primary AML Cells.Cancer Discov. 2017 Jul;7(7):670-672. doi: 10.1158/2159-8290.CD-17-0476. Cancer Discov. 2017. PMID: 28684410
References
-
- Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106 - PubMed
-
- Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–94. - PubMed
-
- Ghiaur G, Wroblewski M, Loges S. Acute myelogenous leukemia and its microenvironment: A Molecular conversation. Semin Hematol. 2015;52:200–6. - PubMed
-
- Pallis M, Turzanski J, Higashi Y, Russell N. P-glycoprotein in acute myeloid leukaemia: therapeutic implications of its association with both a multidrug-resistant and an apoptosis-resistant phenotype. Leuk Lymphoma. 2002;43:1221–8. - PubMed
-
- Buzzai M, Licht JD. New molecular concepts and targets in acute myeloid leukemia. Curr Opin Hematol. 2008;15:82–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
