

Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis
- PMID: 28416670
- PMCID: PMC5465912
- DOI: 10.1073/pnas.1618714114
Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis
Abstract
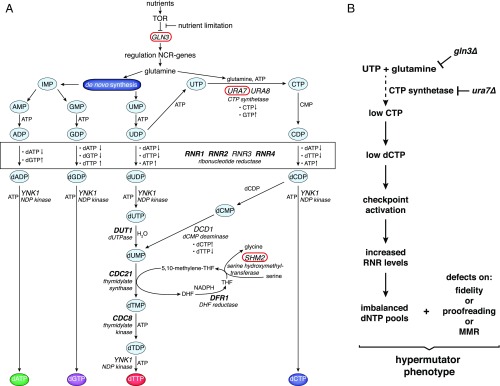
Eukaryotic DNA replication fidelity relies on the concerted action of DNA polymerase nucleotide selectivity, proofreading activity, and DNA mismatch repair (MMR). Nucleotide selectivity and proofreading are affected by the balance and concentration of deoxyribonucleotide (dNTP) pools, which are strictly regulated by ribonucleotide reductase (RNR). Mutations preventing DNA polymerase proofreading activity or MMR function cause mutator phenotypes and consequently increased cancer susceptibility. To identify genes not previously linked to high-fidelity DNA replication, we conducted a genome-wide screen in Saccharomyces cerevisiae using DNA polymerase active-site mutants as a "sensitized mutator background." Among the genes identified in our screen, three metabolism-related genes (GLN3, URA7, and SHM2) have not been previously associated to the suppression of mutations. Loss of either the transcription factor Gln3 or inactivation of the CTP synthetase Ura7 both resulted in the activation of the DNA damage response and imbalanced dNTP pools. Importantly, these dNTP imbalances are strongly mutagenic in genetic backgrounds where DNA polymerase function or MMR activity is partially compromised. Previous reports have shown that dNTP pool imbalances can be caused by mutations altering the allosteric regulation of enzymes involved in dNTP biosynthesis (e.g., RNR or dCMP deaminase). Here, we provide evidence that mutations affecting genes involved in RNR substrate production can cause dNTP imbalances, which cannot be compensated by RNR or other enzymatic activities. Moreover, Gln3 inactivation links nutrient deprivation to increased mutagenesis. Our results suggest that similar genetic interactions could drive mutator phenotypes in cancer cells.
Keywords: CTP biosynthesis; DNA polymerases; DNA replication fidelity; dNTP pool imbalance; mismatch repair.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
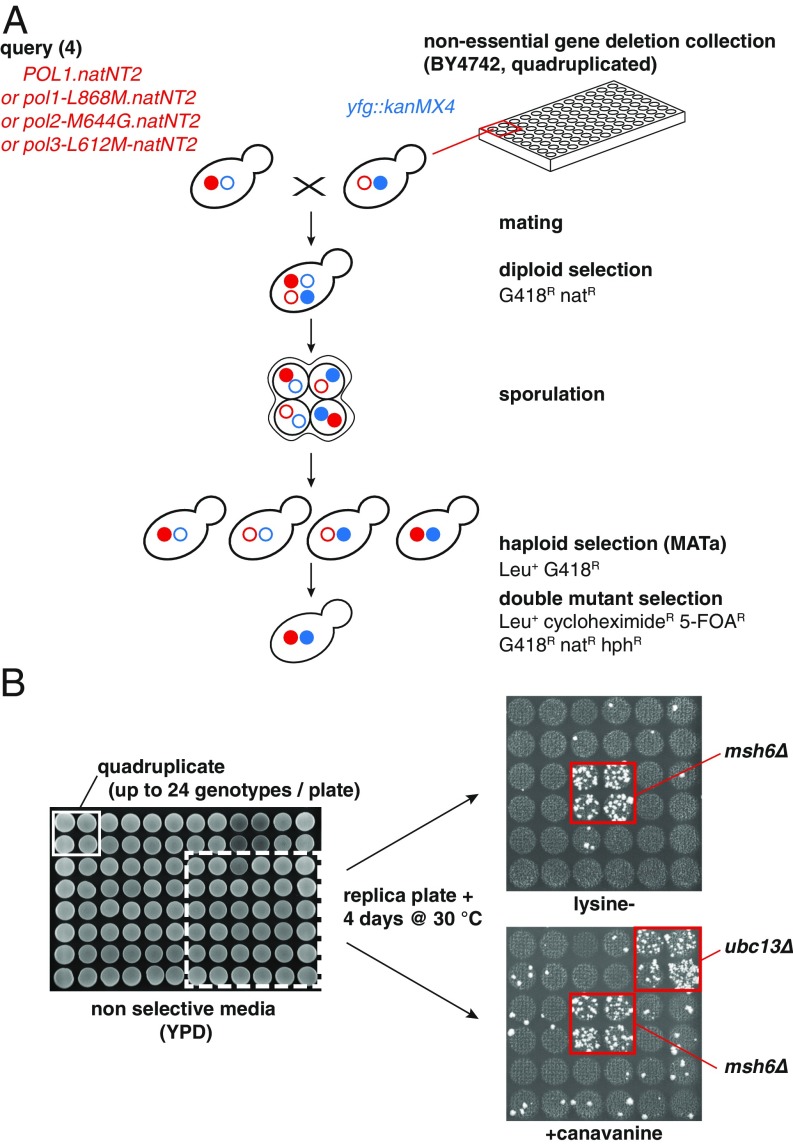

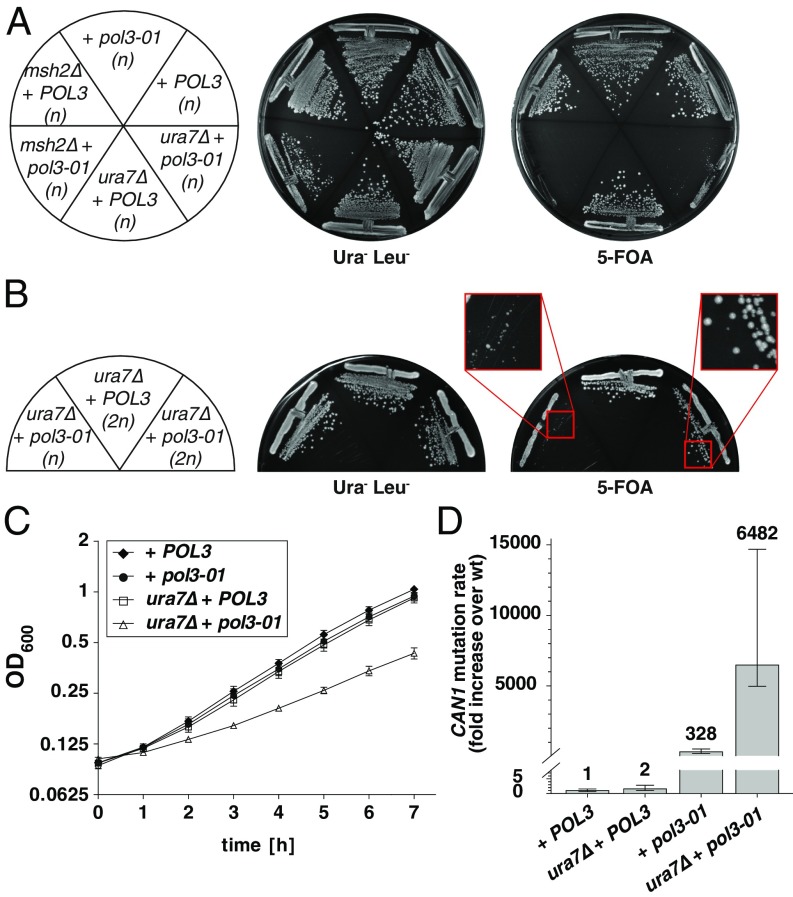
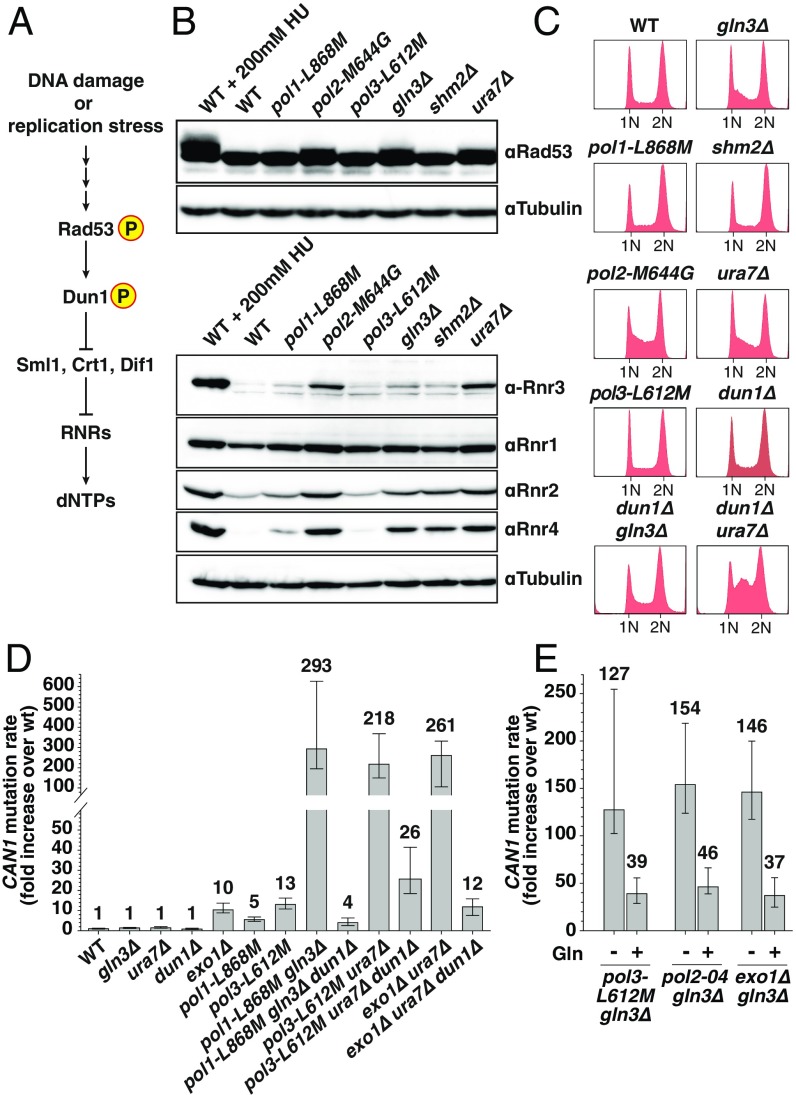
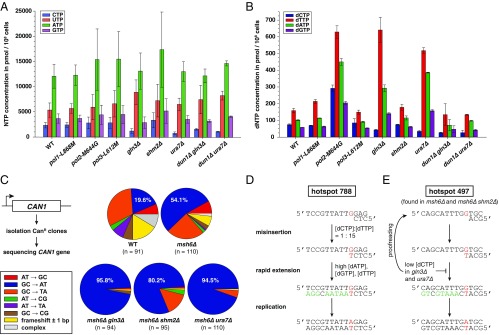
Comment in
-
DNA replication and mismatch repair safeguard against metabolic imbalances.Proc Natl Acad Sci U S A. 2017 May 30;114(22):5561-5563. doi: 10.1073/pnas.1705971114. Epub 2017 May 22. Proc Natl Acad Sci U S A. 2017. PMID: 28533419 Free PMC article. No abstract available.
References
-
- Ganai RA, Johansson E. DNA replication—a matter of fidelity. Mol Cell. 2016;62:745–755. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
