

Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma
- PMID: 28416797
- PMCID: PMC5399304
- DOI: 10.1038/ncomms14920
Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma
Abstract
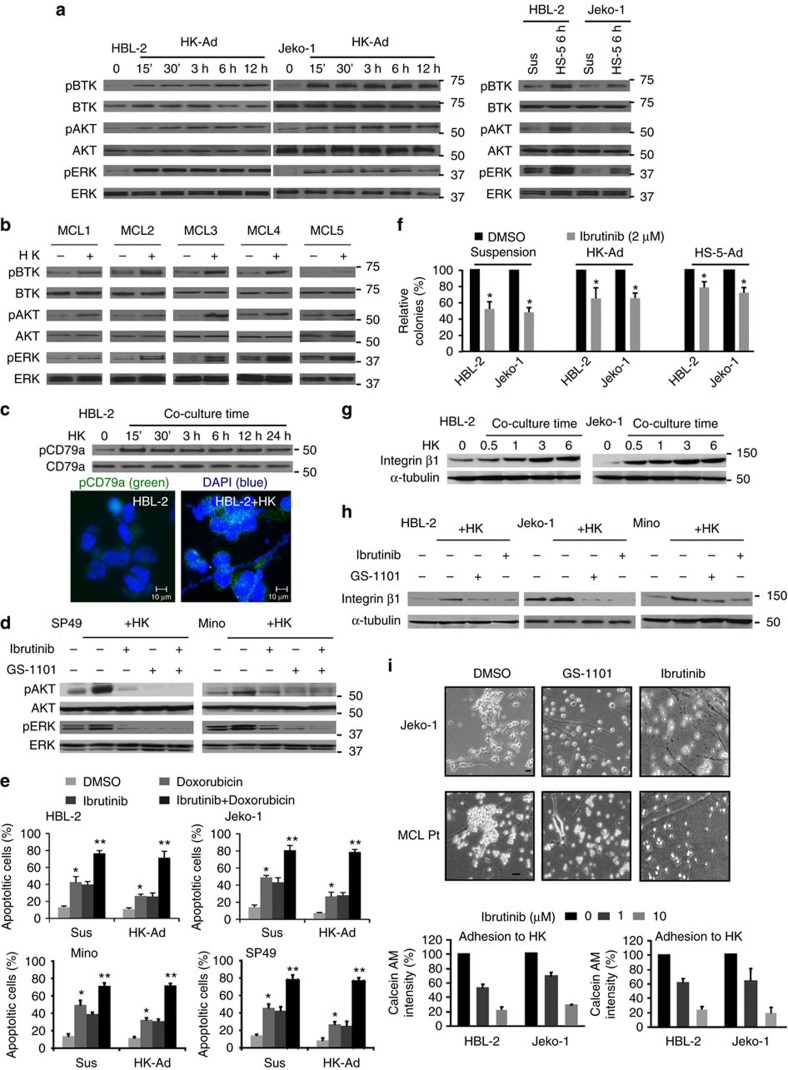
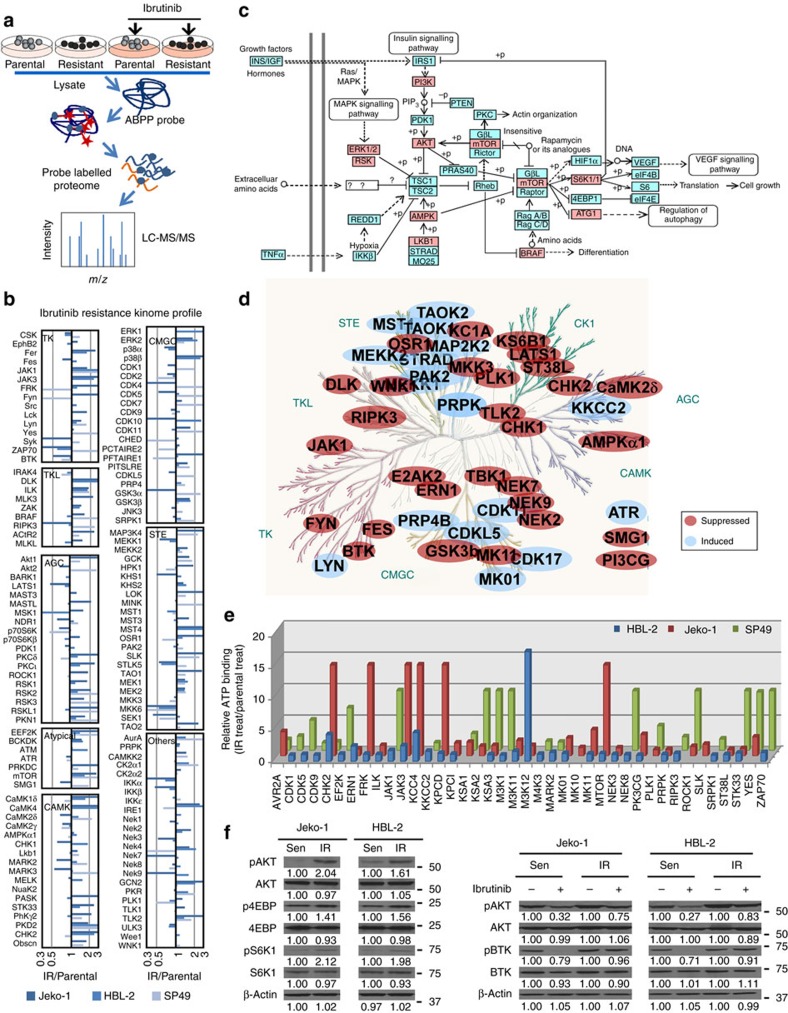
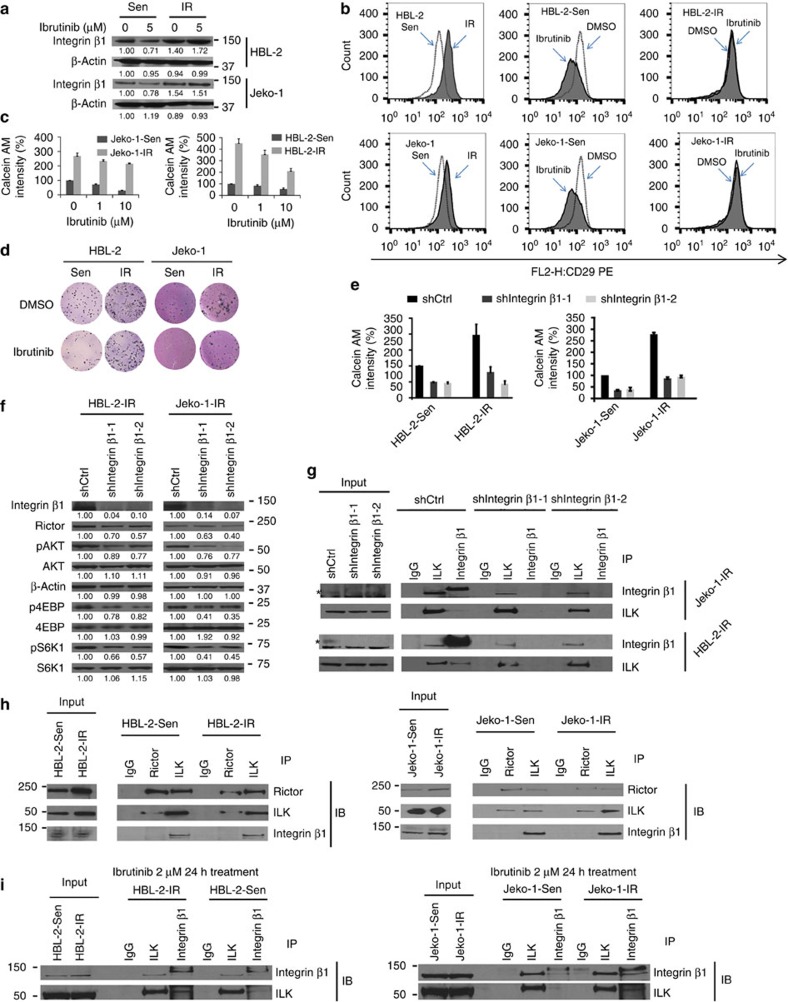
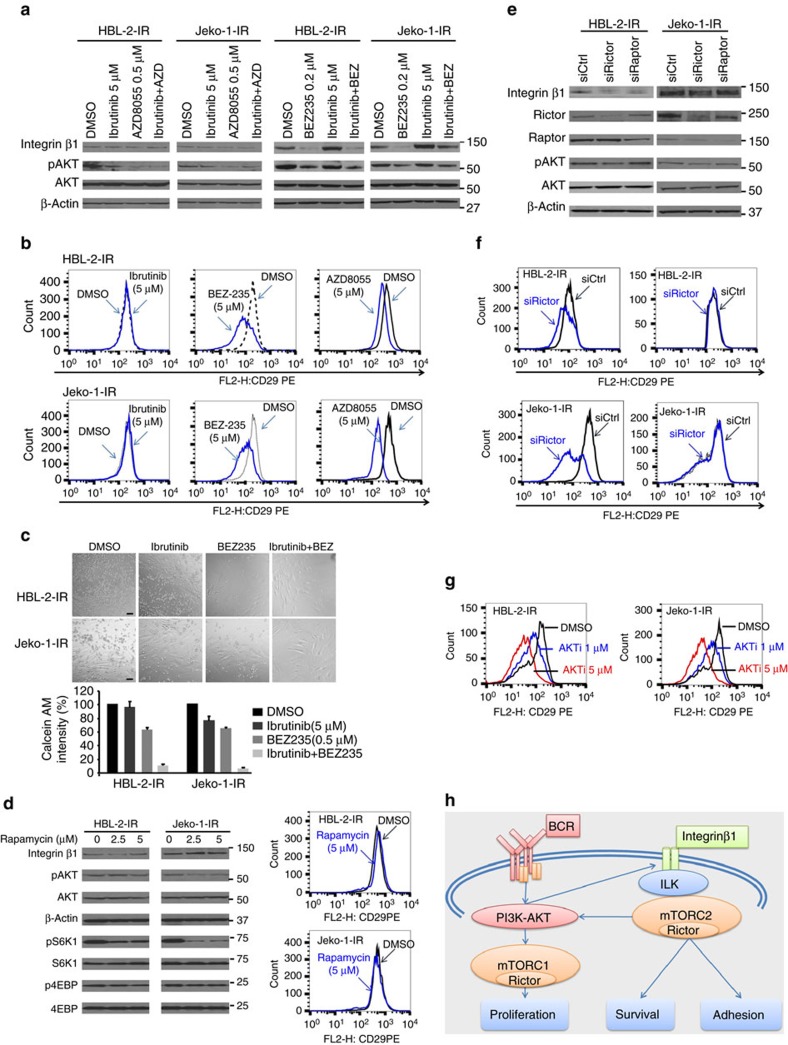
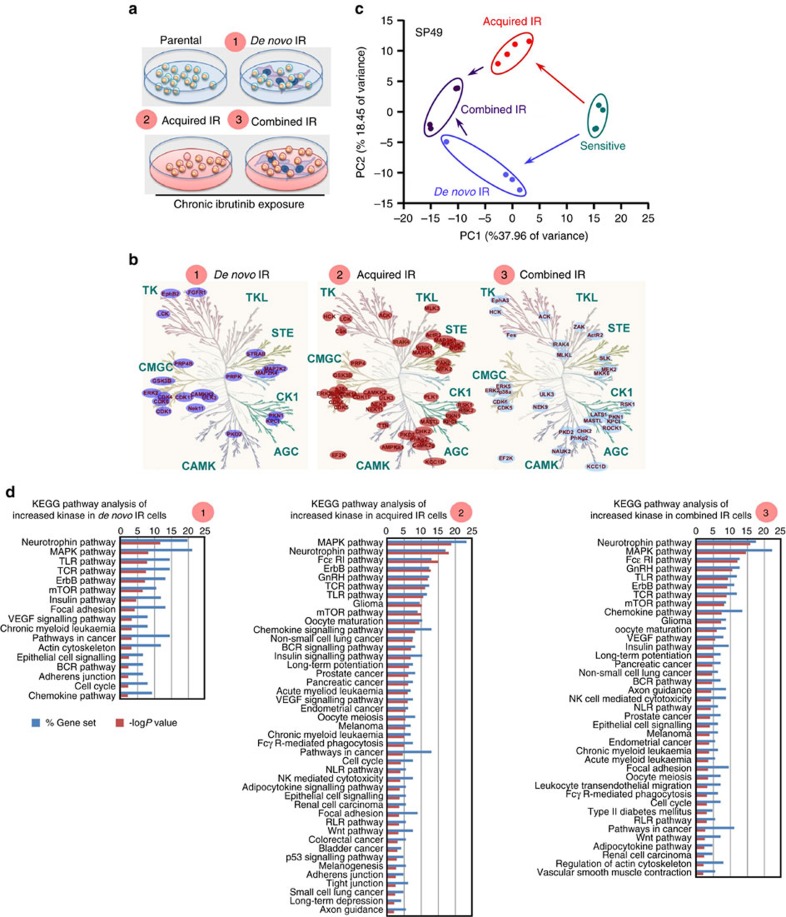
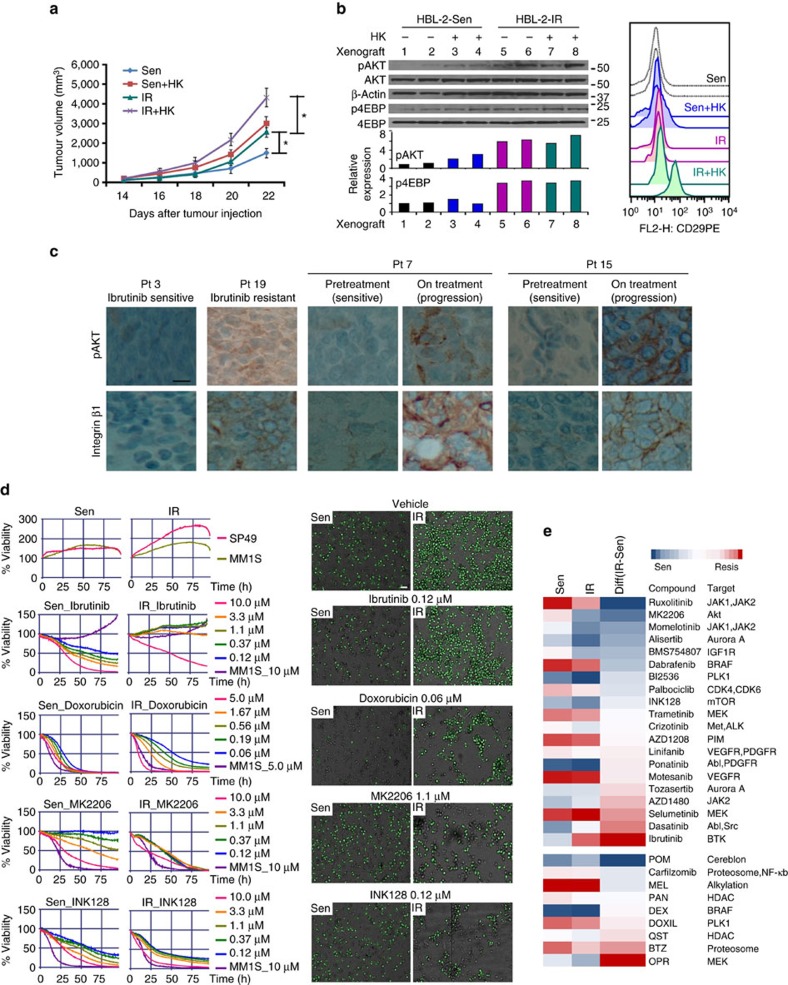
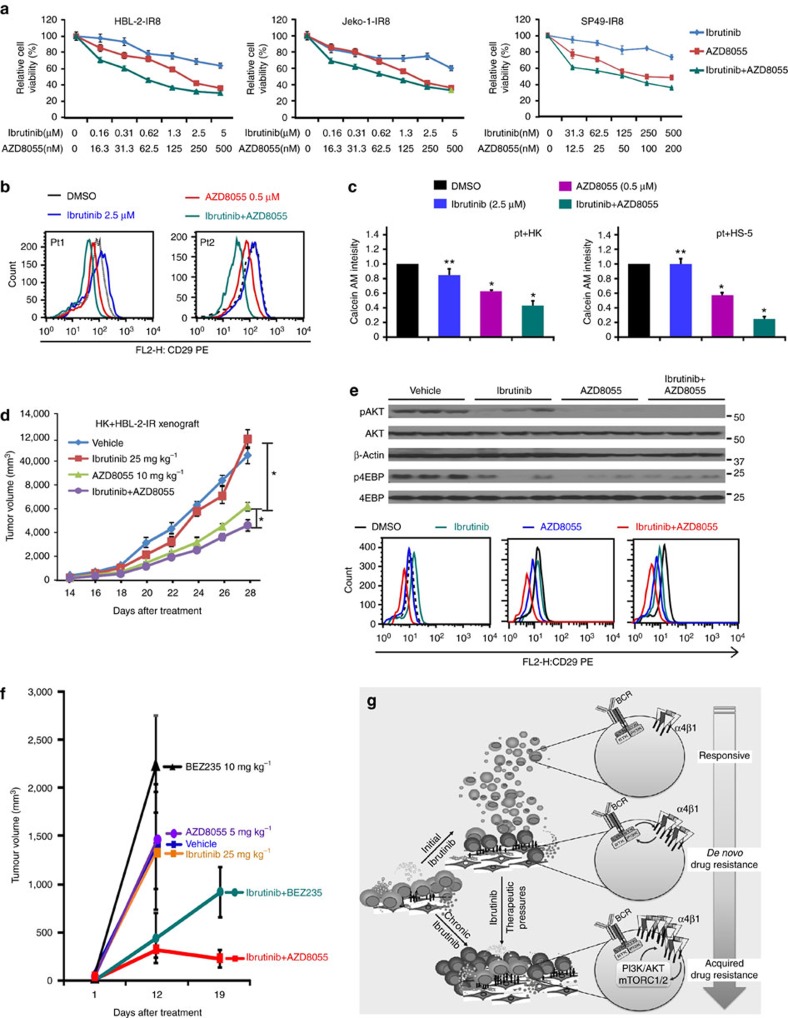
The novel Bruton's tyrosine kinase inhibitor ibrutinib has demonstrated high response rates in B-cell lymphomas; however, a growing number of ibrutinib-treated patients relapse with resistance and fulminant progression. Using chemical proteomics and an organotypic cell-based drug screening assay, we determine the functional role of the tumour microenvironment (TME) in ibrutinib activity and acquired ibrutinib resistance. We demonstrate that MCL cells develop ibrutinib resistance through evolutionary processes driven by dynamic feedback between MCL cells and TME, leading to kinome adaptive reprogramming, bypassing the effect of ibrutinib and reciprocal activation of PI3K-AKT-mTOR and integrin-β1 signalling. Combinatorial disruption of B-cell receptor signalling and PI3K-AKT-mTOR axis leads to release of MCL cells from TME, reversal of drug resistance and enhanced anti-MCL activity in MCL patient samples and patient-derived xenograft models. This study unifies TME-mediated de novo and acquired drug resistance mechanisms and provides a novel combination therapeutic strategy against MCL and other B-cell malignancies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma.Oncogene. 2019 Mar;38(11):1802-1814. doi: 10.1038/s41388-018-0550-3. Epub 2018 Oct 25. Oncogene. 2019. PMID: 30361685
-
Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib.Blood. 2015 Sep 24;126(13):1565-74. doi: 10.1182/blood-2015-04-639542. Epub 2015 Aug 7. Blood. 2015. PMID: 26254443 Free PMC article.
-
Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma.Cancer Discov. 2014 Sep;4(9):1022-35. doi: 10.1158/2159-8290.CD-14-0098. Epub 2014 Jul 31. Cancer Discov. 2014. PMID: 25082755 Free PMC article.
-
Ibrutinib for mantle cell lymphoma.Future Oncol. 2016 Feb;12(4):477-91. doi: 10.2217/fon.15.342. Epub 2016 Jan 13. Future Oncol. 2016. PMID: 26759179 Review.
-
Ibrutinib resistance in mantle cell lymphoma: clinical, molecular and treatment aspects.Br J Haematol. 2018 May;181(3):306-319. doi: 10.1111/bjh.15108. Epub 2018 Jan 23. Br J Haematol. 2018. PMID: 29359797 Review.
Cited by
-
Ibrutinib in Chronic Lymphocytic Leukemia: Clinical Applications, Drug Resistance, and Prospects.Onco Targets Ther. 2020 May 29;13:4877-4892. doi: 10.2147/OTT.S249586. eCollection 2020. Onco Targets Ther. 2020. PMID: 32581549 Free PMC article. Review.
-
Resistance to BTK inhibition by ibrutinib can be overcome by preventing FOXO3a nuclear export and PI3K/AKT activation in B-cell lymphoid malignancies.Cell Death Dis. 2019 Dec 4;10(12):924. doi: 10.1038/s41419-019-2158-0. Cell Death Dis. 2019. PMID: 31801949 Free PMC article.
-
Novel method to identify group-specific non-catalytic pockets of human kinome for drug design.RSC Adv. 2020 Jan 10;10(4):2004-2015. doi: 10.1039/c9ra07471f. eCollection 2020 Jan 8. RSC Adv. 2020. PMID: 35494619 Free PMC article.
-
Ibrutinib resistance in a patient with transformed diffuse large B-cell lymphoma from primary pulmonary mucosa-associated lymphoid tissue lymphoma.Cancer Biol Ther. 2020 Apr 2;21(4):303-308. doi: 10.1080/15384047.2019.1700743. Epub 2020 Jan 13. Cancer Biol Ther. 2020. PMID: 31931656 Free PMC article.
-
Adaptive chromatin remodeling and transcriptional changes of the functional kinome in tumor cells in response to targeted kinase inhibition.J Biol Chem. 2022 Feb;298(2):101525. doi: 10.1016/j.jbc.2021.101525. Epub 2021 Dec 24. J Biol Chem. 2022. PMID: 34958800 Free PMC article. Review.
References
-
- Shipp M. A. BCR signalling and survival pathways in lymphoma. Hematol. Oncol. 33, 140–141 (2015).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
