

Pathogenic variants that alter protein code often disrupt splicing
- PMID: 28416821
- PMCID: PMC6679692
- DOI: 10.1038/ng.3837
Pathogenic variants that alter protein code often disrupt splicing
Abstract
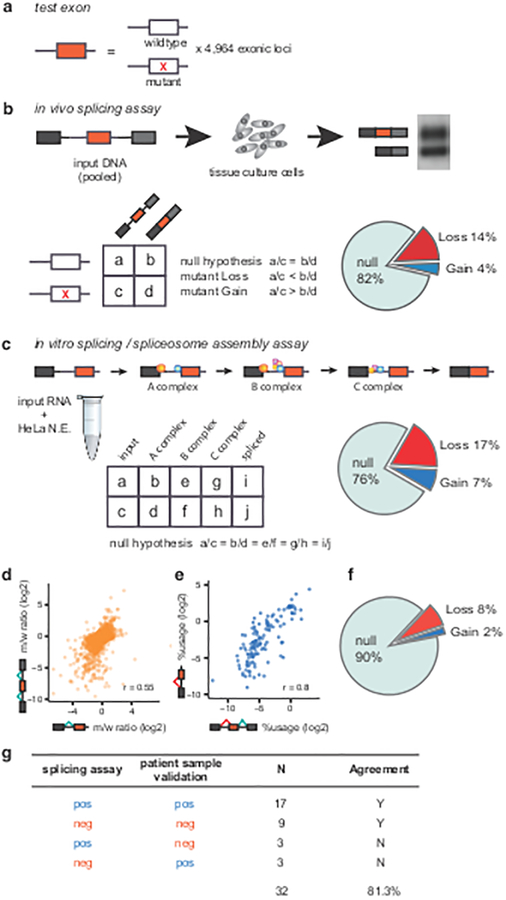
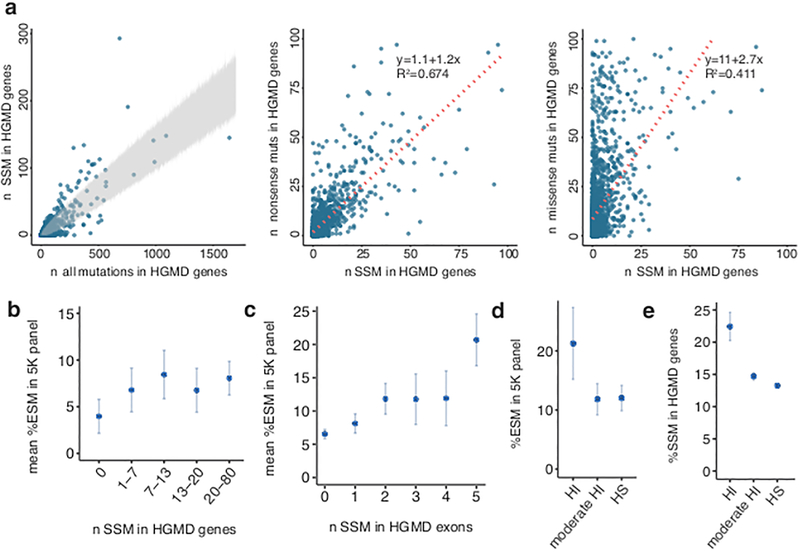
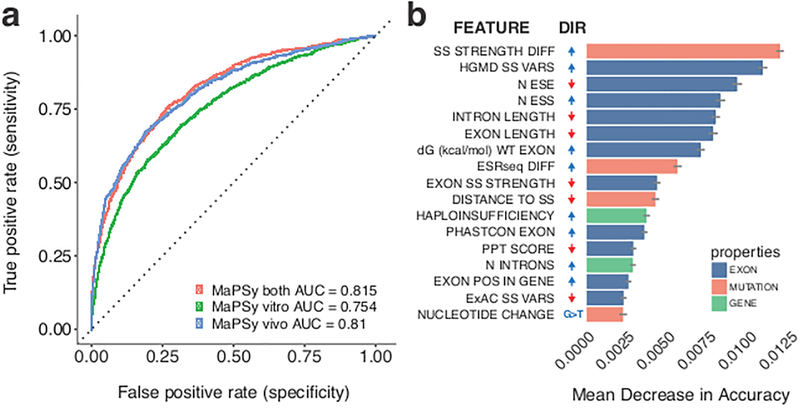
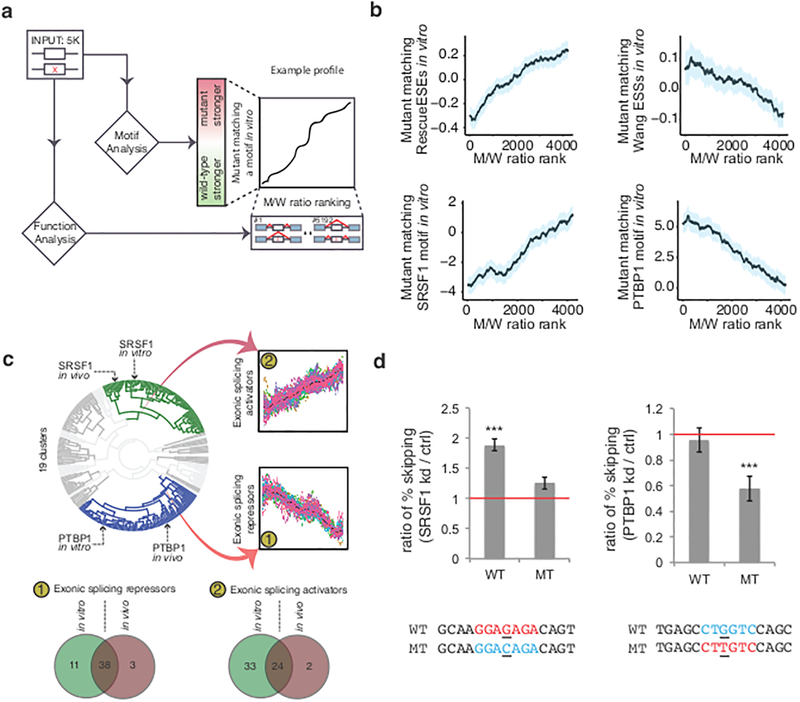
The lack of tools to identify causative variants from sequencing data greatly limits the promise of precision medicine. Previous studies suggest that one-third of disease-associated alleles alter splicing. We discovered that the alleles causing splicing defects cluster in disease-associated genes (for example, haploinsufficient genes). We analyzed 4,964 published disease-causing exonic mutations using a massively parallel splicing assay (MaPSy), which showed an 81% concordance rate with splicing in patient tissue. Approximately 10% of exonic mutations altered splicing, mostly by disrupting multiple stages of spliceosome assembly. We present a large-scale characterization of exonic splicing mutations using a new technology that facilitates variant classification and keeps pace with variant discovery.
Conflict of interest statement
Competing Financial Interests
The authors declare no competing financial interests.
Figures
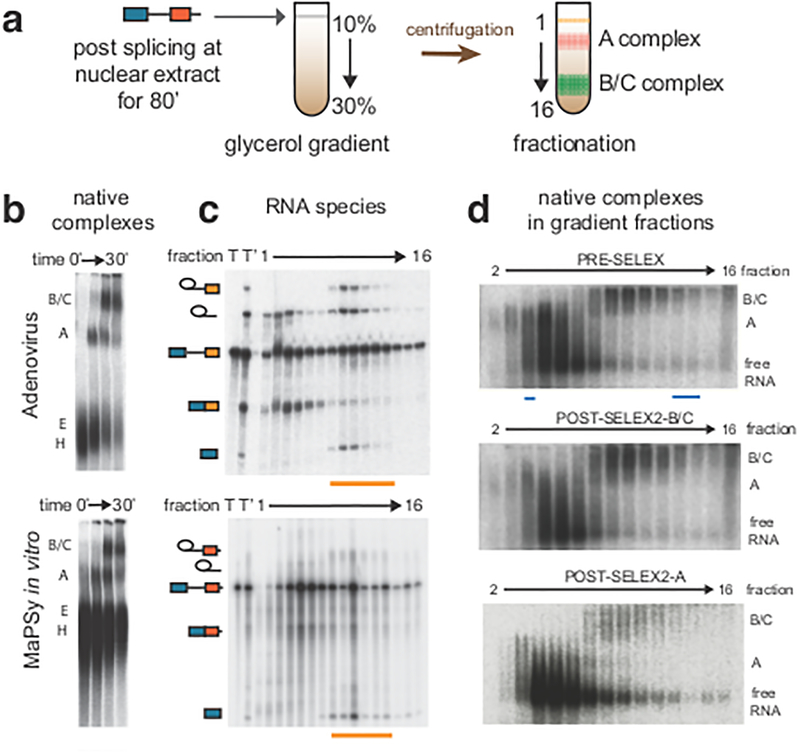
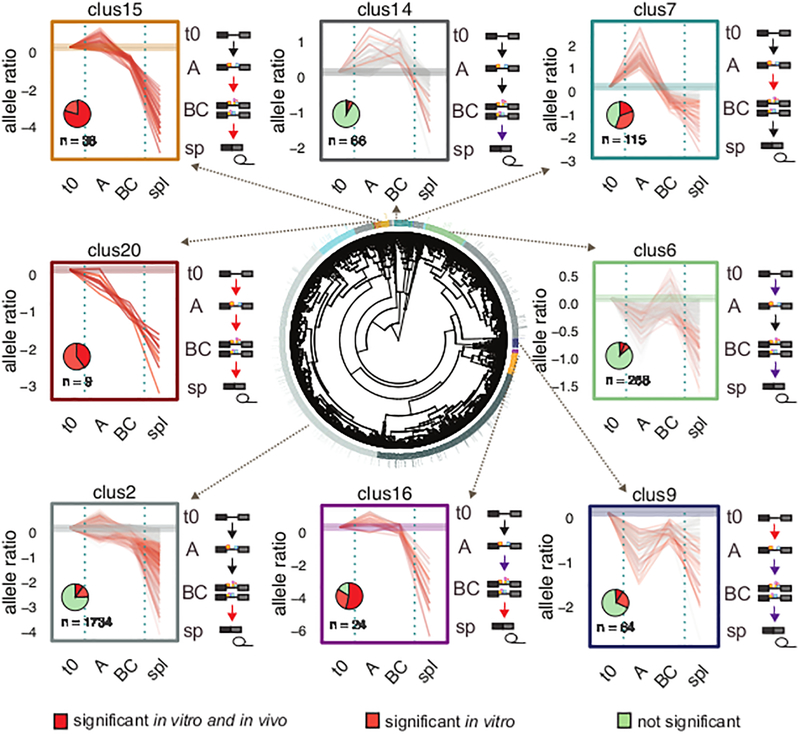
References
-
- Bamshad MJ et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12, 745–55 (2011). - PubMed
Online Method References
-
- Yeo G & Burge CB Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal ofComputationaI Biology 11, 377–94 (2004). - PubMed
-
- Kursa MB, Jankowski A & Rudnicki WR Boruta - A System for Feature Selection. Fundamenta Informaticae 101, 271–286 (2010).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
