

Atypical juvenile presentation of GM2 gangliosidosis AB in a patient compound-heterozygote for c.259G > T and c.164C > T mutations in the GM2A gene
- PMID: 28417072
- PMCID: PMC5388932
- DOI: 10.1016/j.ymgmr.2017.01.017
Atypical juvenile presentation of GM2 gangliosidosis AB in a patient compound-heterozygote for c.259G > T and c.164C > T mutations in the GM2A gene
Abstract
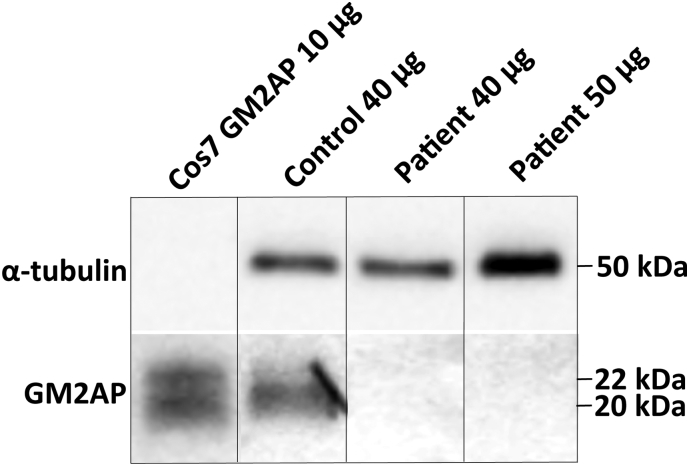
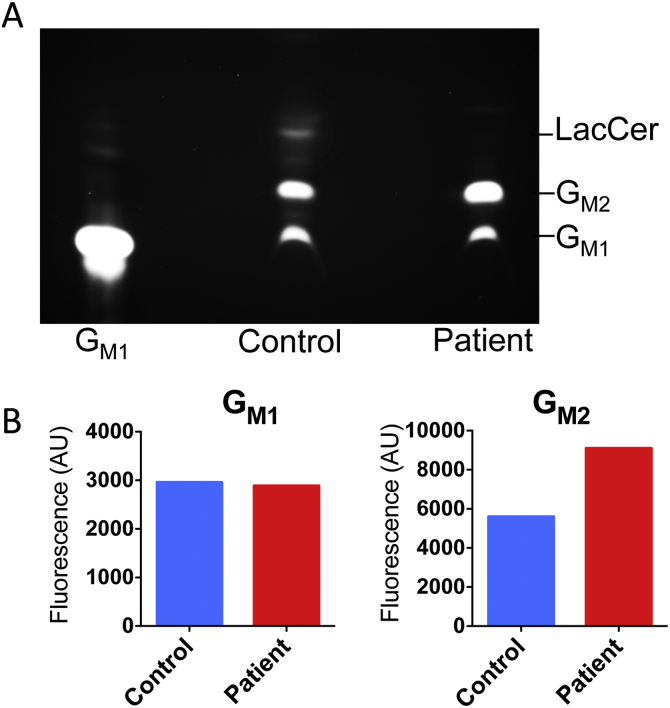
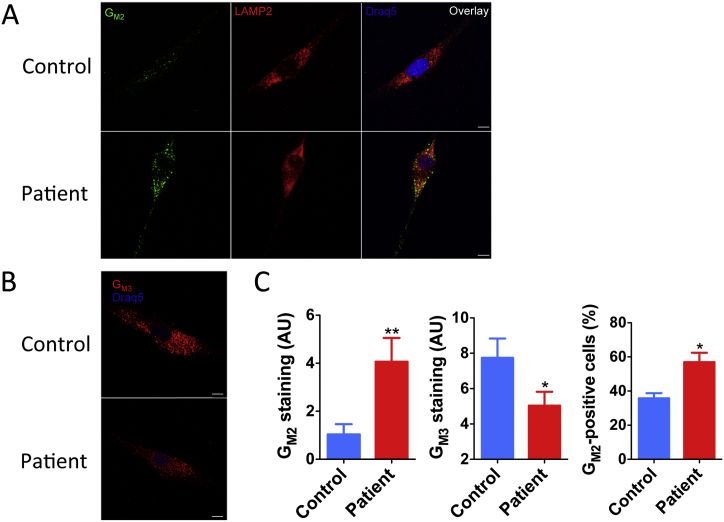
GM2-gangliosidosis, AB variant is an extremely rare autosomal recessive inherited disorder caused by mutations in the GM2A gene that encodes GM2 ganglioside activator protein (GM2AP). GM2AP is necessary for solubilisation of GM2 ganglioside in endolysosomes and its presentation to β-hexosaminidase A. Conversely GM2AP deficiency impairs lysosomal catabolism of GM2 ganglioside, leading to its storage in cells and tissues. We describe a 9-year-old child with an unusual juvenile clinical onset of GM2-gangliosidosis AB. At the age of 3 years he presented with global developmental delay, progressive epilepsy, intellectual disability, axial hypertonia, spasticity, seizures and ataxia, but without the macular cherry-red spots typical for GM2 gangliosidosis. Brain MRI detected a rapid onset of diffuse atrophy, whereas whole exome sequencing showed that the patient is a compound heterozygote for two mutations in GM2A: a novel nonsense mutation, c.259G > T (p.E87X) and a missense mutation c.164C > T (p.P55L) that was recently identified in homozygosity in patients of a Saudi family with a progressive chorea-dementia syndrome. Western blot analysis showed an absence of GM2AP in cultured fibroblasts from the patient, suggesting that both mutations interfere with the synthesis and/or folding of the protein. Finally, impaired catabolism of GM2 ganglioside in the patient's fibroblasts was demonstrated by metabolic labeling with fluorescently labeled GM1 ganglioside and by immunohistochemistry with anti-GM2 and anti-GM3 antibodies. Our observation expands the molecular and clinical spectrum of molecular defects linked to GM2-gangliosidosis and suggests novel diagnostic approach by whole exome sequencing and perhaps ganglioside analysis in cultured patient's cells.
Keywords: GM2 ganglioside; GM2 ganglioside activator protein; Gangliosidosis; Lysosomal storage disease.
Figures
References
-
- Burkhardt J.K., Huttler S., Klein A., Mobius W., Habermann A., Griffiths G., Sandhoff K. Accumulation of sphingolipids in SAP-precursor (prosaposin)-deficient fibroblasts occurs as intralysosomal membrane structures and can be completely reversed by treatment with human SAP-precursor. Eur. J. Cell Biol. 1997;73:10–18. - PubMed
-
- Mobius W., Herzog V., Sandhoff K., Schwarzmann G. Intracellular distribution of a biotin-labeled ganglioside, GM1, by immunoelectron microscopy after endocytosis in fibroblasts. J. Histochem. Cytochem. 1999;47:1005–1014. - PubMed
-
- Gallala H.D., Sandhoff K. Biological function of the cellular lipid BMP-BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem. Res. 2011;36:1594–1600. - PubMed
-
- Gallala H.D., Breiden B., Sandhoff K. Regulation of the NPC2 protein-mediated cholesterol trafficking by membrane lipids. J. Neurochem. 2011;116:702–707. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
