

Predicting Protein-protein Association Rates using Coarse-grained Simulation and Machine Learning
- PMID: 28418043
- PMCID: PMC5394550
- DOI: 10.1038/srep46622
Predicting Protein-protein Association Rates using Coarse-grained Simulation and Machine Learning
Abstract
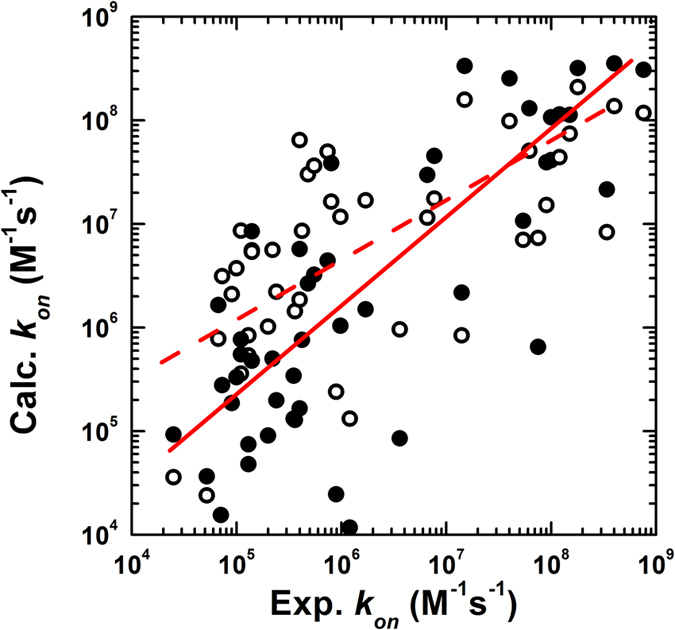
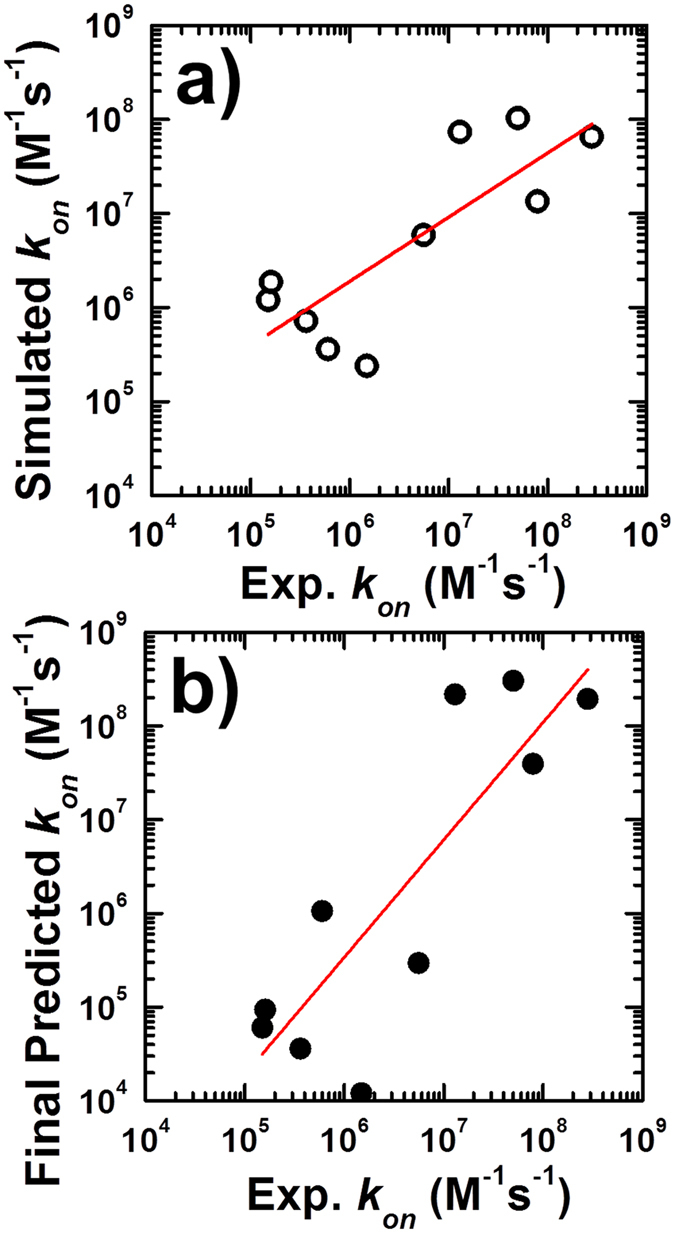
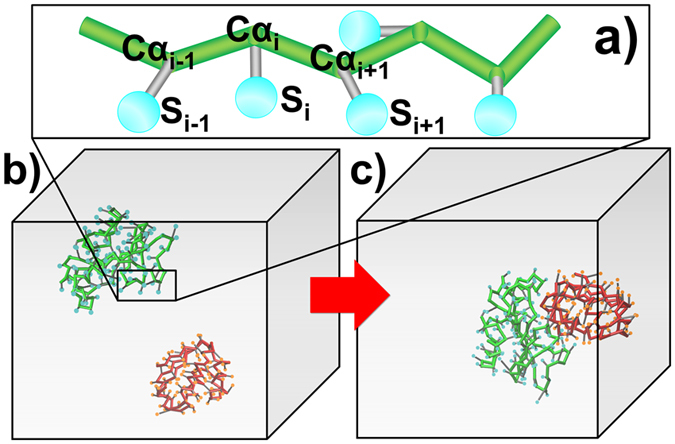
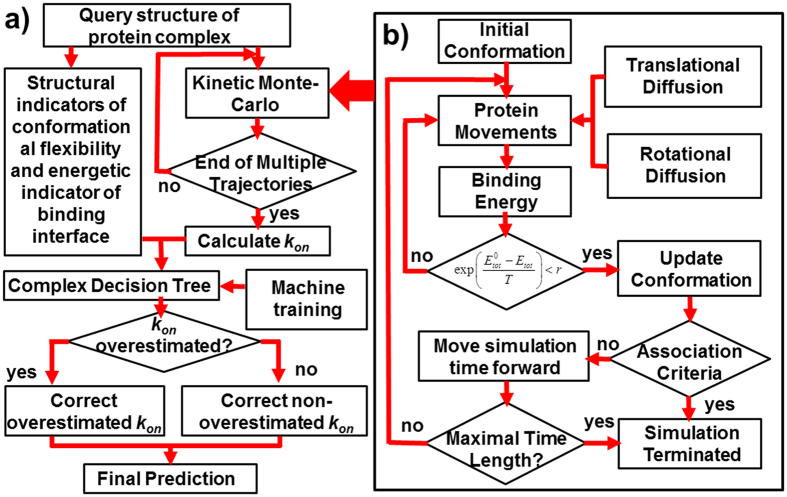
Protein-protein interactions dominate all major biological processes in living cells. We have developed a new Monte Carlo-based simulation algorithm to study the kinetic process of protein association. We tested our method on a previously used large benchmark set of 49 protein complexes. The predicted rate was overestimated in the benchmark test compared to the experimental results for a group of protein complexes. We hypothesized that this resulted from molecular flexibility at the interface regions of the interacting proteins. After applying a machine learning algorithm with input variables that accounted for both the conformational flexibility and the energetic factor of binding, we successfully identified most of the protein complexes with overestimated association rates and improved our final prediction by using a cross-validation test. This method was then applied to a new independent test set and resulted in a similar prediction accuracy to that obtained using the training set. It has been thought that diffusion-limited protein association is dominated by long-range interactions. Our results provide strong evidence that the conformational flexibility also plays an important role in regulating protein association. Our studies provide new insights into the mechanism of protein association and offer a computationally efficient tool for predicting its rate.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
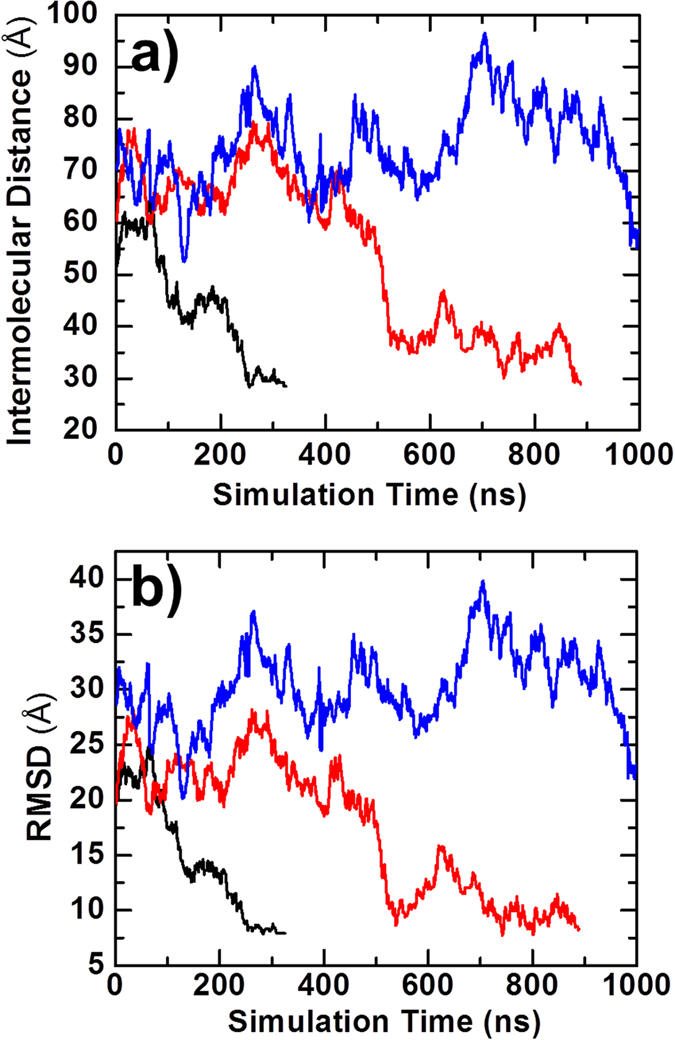
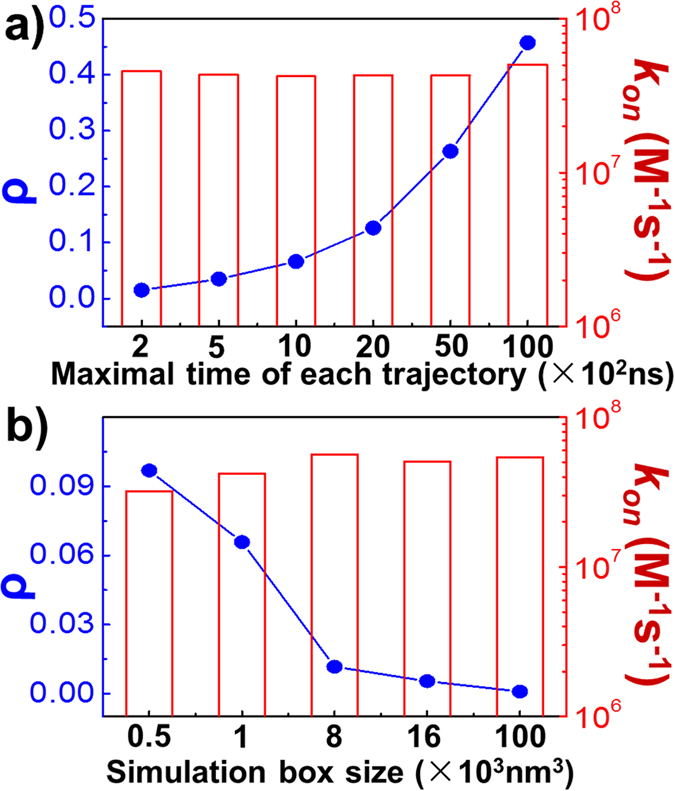
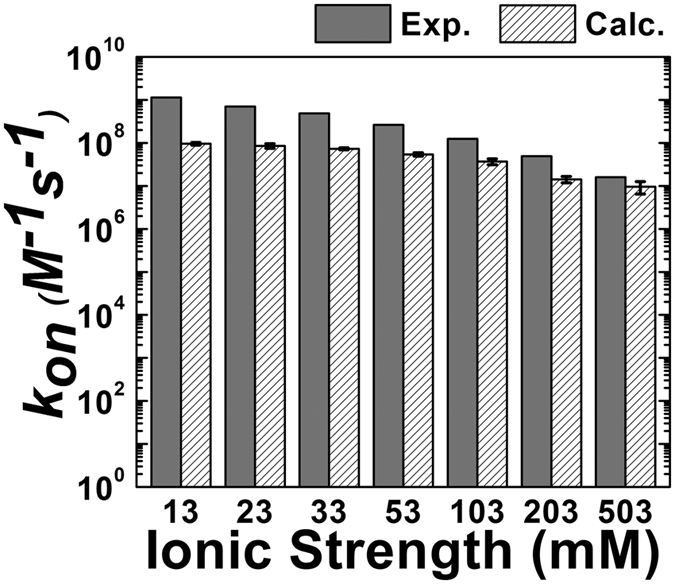
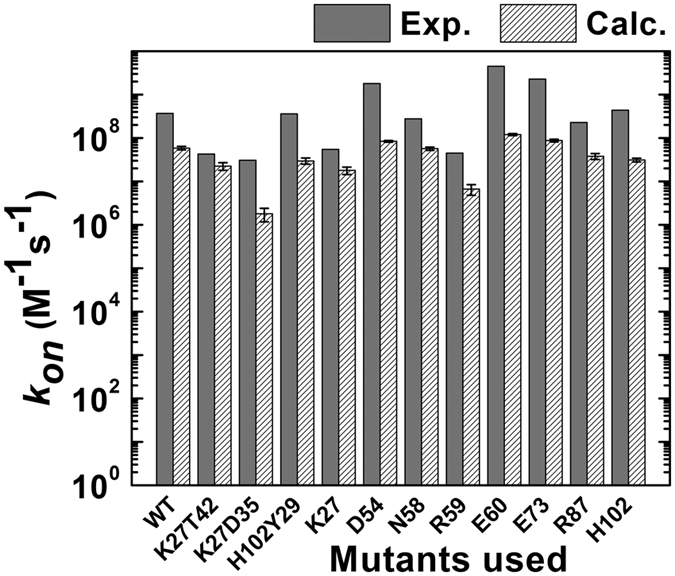
References
-
- Janin J. & Chothia C. The structure of protein-protein recognition sites. J Biol Chem 265, 16027–16030 (1990). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
