

Contributions of MET activation to BCR-ABL1 tyrosine kinase inhibitor resistance in chronic myeloid leukemia cells
- PMID: 28418880
- PMCID: PMC5503566
- DOI: 10.18632/oncotarget.16314
Contributions of MET activation to BCR-ABL1 tyrosine kinase inhibitor resistance in chronic myeloid leukemia cells
Abstract
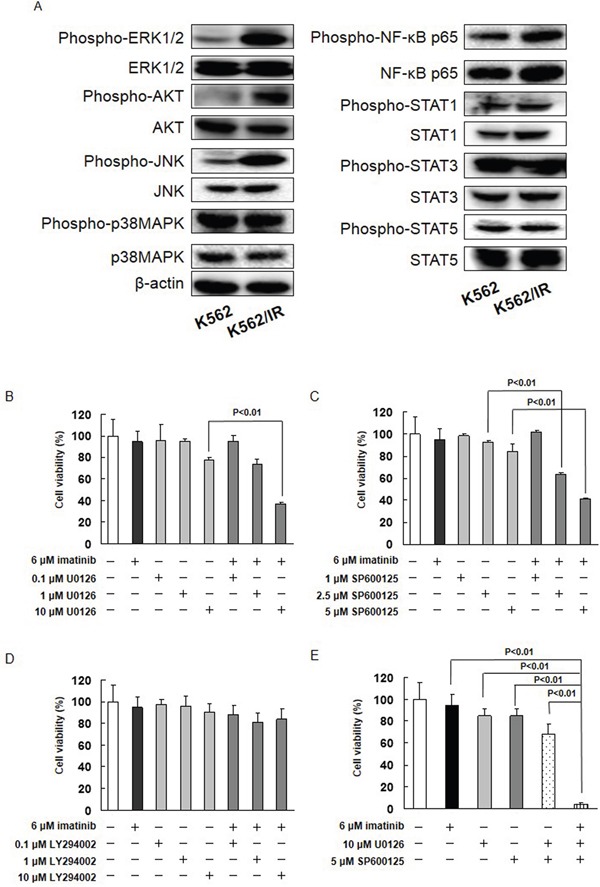
Resistance to the breakpoint cluster region-abelson 1 (BCR-ABL1) tyrosine kinase inhibitor (TKI) imatinib poses a major problem when treating chronic myeloid leukemia (CML). Imatinib resistance often results from a secondary mutation in BCR-ABL1. However, in the absence of a mutation in BCR-ABL1, the basis of BCR-ABL1-independent resistance must be elucidated. To gain insight into the mechanisms of BCR-ABL1-independent imatinib resistance, we performed an array-based comparative genomic hybridization. We identified various resistance-related genes, and focused on MET. Treatment with a MET inhibitor resensitized K562/IR cells to BCR-ABL1 TKIs. Combined treatment of K562/IR cells with imatinib and a MET inhibitor suppressed extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK) activation, but did not affect AKT activation. Our findings implicate the MET/ERK and MET/JNK pathways in conferring resistance to imatinib, providing new insights into the mechanisms of BCR-ABL1 TKI resistance in CML.
Keywords: ERK1/2; JNK; MET; chronic myeloid leukemia; imatinib resistance.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures




Similar articles
-
Hypoxia-inducible factor 1α inhibitor induces cell death via suppression of BCR-ABL1 and Met expression in BCR-ABL1 tyrosine kinase inhibitor sensitive and resistant chronic myeloid leukemia cells.BMB Rep. 2023 Feb;56(2):78-83. doi: 10.5483/BMBRep.2022-0095. BMB Rep. 2023. PMID: 36195570 Free PMC article.
-
[MET/ERK and MET/JNK Pathway Activation Is Involved in BCR-ABL Inhibitor-resistance in Chronic Myeloid Leukemia].Yakugaku Zasshi. 2018;138(12):1461-1466. doi: 10.1248/yakushi.18-00142. Yakugaku Zasshi. 2018. PMID: 30504658 Review. Japanese.
-
A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia.Leukemia. 2016 Jul;30(7):1493-501. doi: 10.1038/leu.2016.51. Epub 2016 Mar 8. Leukemia. 2016. PMID: 27044711 Free PMC article.
-
The novel anticancer agent JNJ-26854165 is active in chronic myeloid leukemic cells with unmutated BCR/ABL and T315I mutant BCR/ABL through promoting proteosomal degradation of BCR/ABL proteins.Oncotarget. 2017 Jan 31;8(5):7777-7790. doi: 10.18632/oncotarget.13951. Oncotarget. 2017. PMID: 27999193 Free PMC article.
-
New Insights into the Molecular Resistance Mechanisms of Chronic Myeloid Leukemia.Curr Cancer Drug Targets. 2016;16(4):323-45. doi: 10.2174/1568009615666150921141004. Curr Cancer Drug Targets. 2016. PMID: 26391311 Review.
Cited by
-
Hypoxia-inducible factor 1α inhibitor induces cell death via suppression of BCR-ABL1 and Met expression in BCR-ABL1 tyrosine kinase inhibitor sensitive and resistant chronic myeloid leukemia cells.BMB Rep. 2023 Feb;56(2):78-83. doi: 10.5483/BMBRep.2022-0095. BMB Rep. 2023. PMID: 36195570 Free PMC article.
-
Overactivation of Akt Contributes to MEK Inhibitor Primary and Acquired Resistance in Colorectal Cancer Cells.Cancers (Basel). 2019 Nov 25;11(12):1866. doi: 10.3390/cancers11121866. Cancers (Basel). 2019. PMID: 31769426 Free PMC article.
-
The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells.Int J Mol Sci. 2020 Oct 24;21(21):7905. doi: 10.3390/ijms21217905. Int J Mol Sci. 2020. PMID: 33114380 Free PMC article.
-
The acquisition order of leukemic drug resistance mutations is directed by the selective fitness associated with each resistance mechanism.Sci Rep. 2023 Aug 11;13(1):13110. doi: 10.1038/s41598-023-40279-2. Sci Rep. 2023. PMID: 37567965 Free PMC article.
-
MDM2 inhibitors induce apoptosis by suppressing MDM2 and enhancing p53, Bax, Puma and Noxa expression levels in imatinib‑resistant chronic myeloid leukemia cells.Biomed Rep. 2025 Feb 12;22(4):65. doi: 10.3892/br.2025.1943. eCollection 2025 Apr. Biomed Rep. 2025. PMID: 39991005 Free PMC article.
References
-
- Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. - PubMed
-
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. - PubMed
-
- Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. - PubMed
-
- Tomoda K, Kato JY, Tatsumi E, Takahashi T, Matsuo Y, Yoneda-Kato N. The Jab1/COP9 signalosome subcomplex is a downstream mediator of Bcr-Abl kinase activity and facilitates cell-cycle progression. Blood. 2005;105:775–783. - PubMed
-
- von Bubnoff N, Peschel C, Duyster J. Resistance of Philadelphia-chromosome positive leukemia towards the kinase inhibitor imatinib (STI571, Glivec): a targeted oncoprotein strikes back. Leukemia. 2003;17:829–838. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
