

A membrane-embedded pathway delivers general anesthetics to two interacting binding sites in the Gloeobacter violaceus ion channel
- PMID: 28420728
- PMCID: PMC5465477
- DOI: 10.1074/jbc.M117.780197
A membrane-embedded pathway delivers general anesthetics to two interacting binding sites in the Gloeobacter violaceus ion channel
Abstract
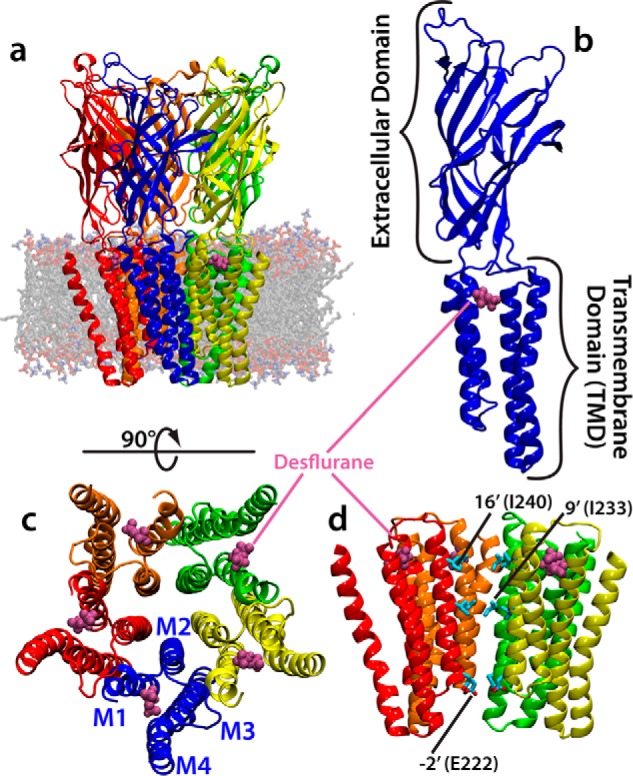
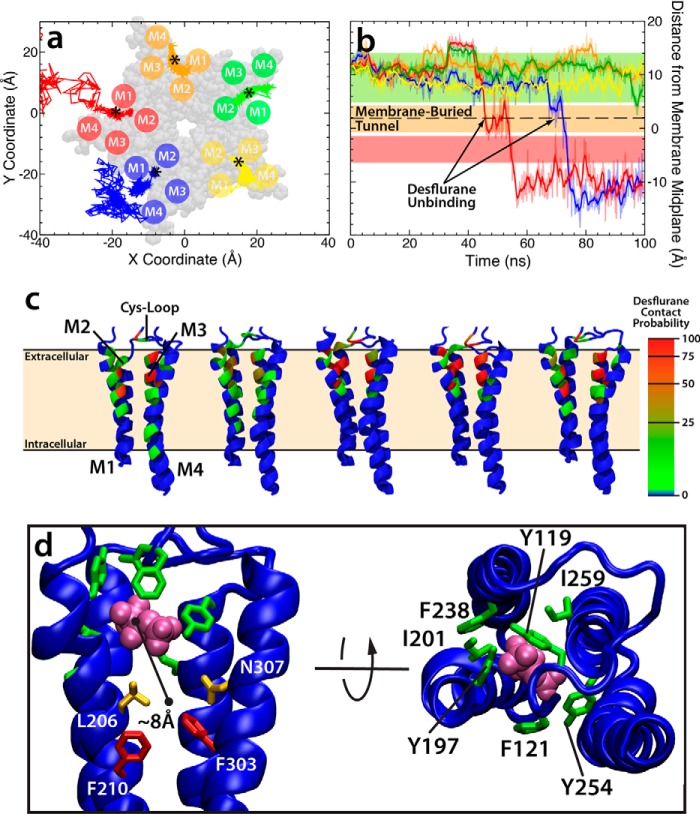
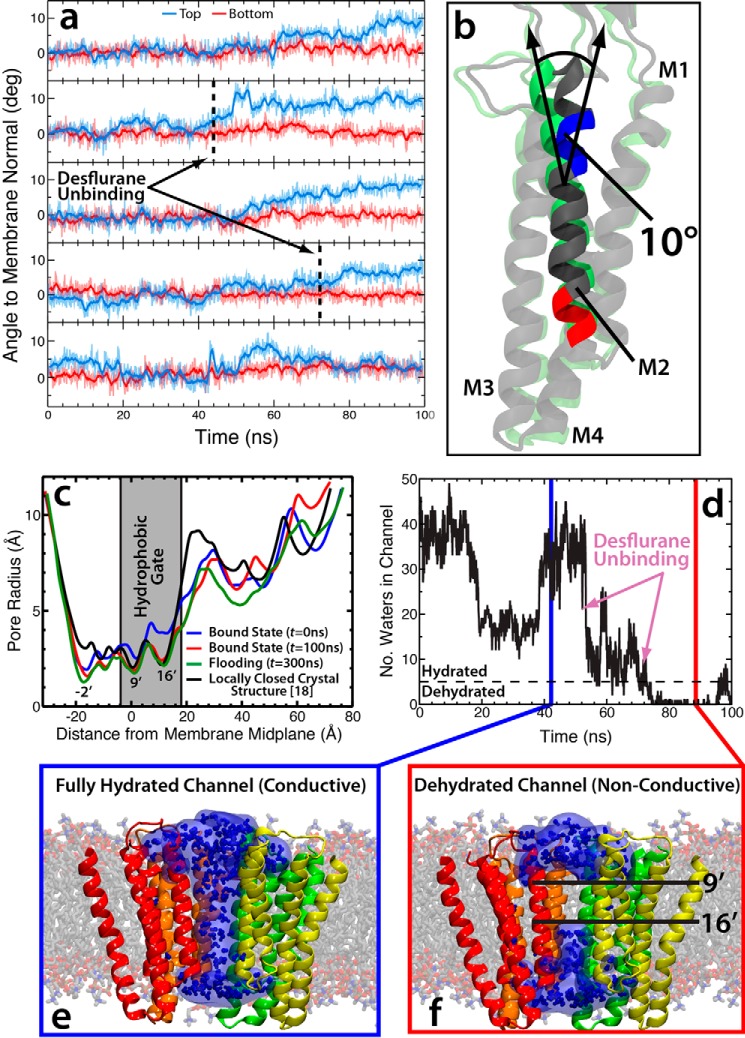
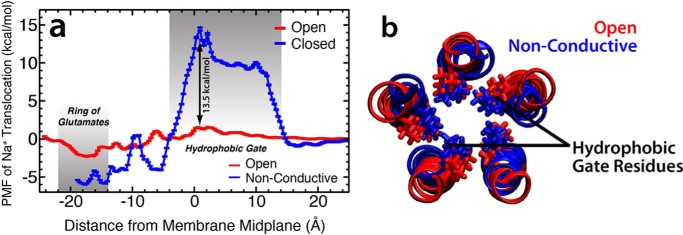
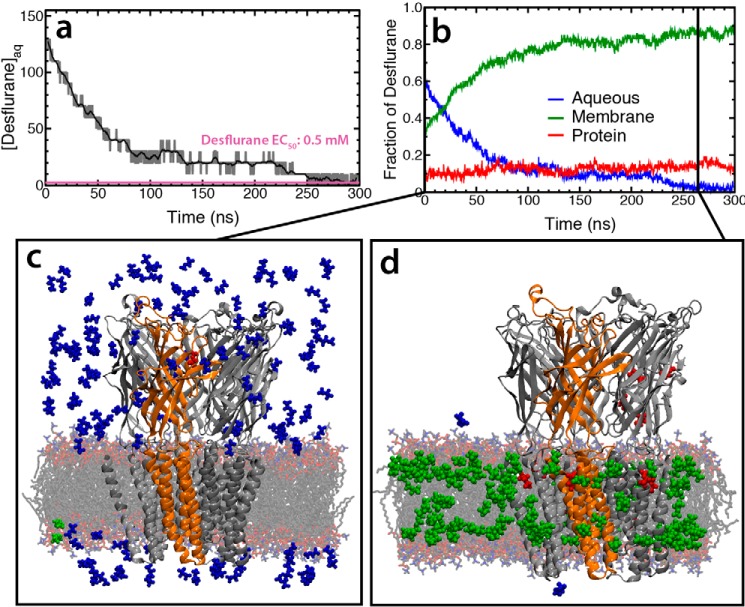
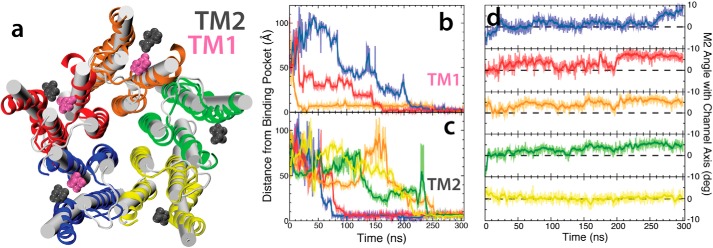
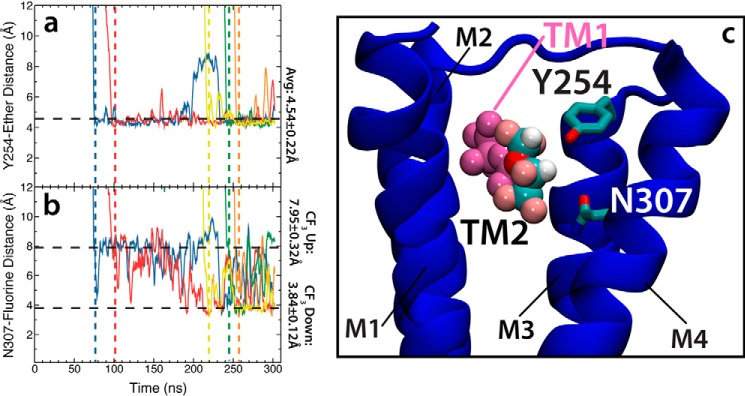
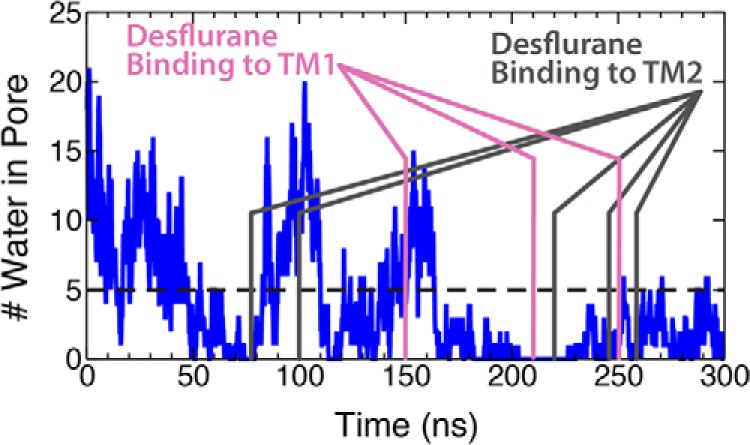
General anesthetics exert their effects on the central nervous system by acting on ion channels, most notably pentameric ligand-gated ion channels. Although numerous studies have focused on pentameric ligand-gated ion channels, the details of anesthetic binding and channel modulation are still debated. A better understanding of the anesthetic mechanism of action is necessary for the development of safer and more efficacious drugs. Herein, we present a computational study identifying two anesthetic binding sites in the transmembrane domain of the Gloeobacter violaceus ligand-gated ion channel (GLIC) channel, characterize the putative binding pathway, and observe structural changes associated with channel function. Molecular simulations of desflurane reveal a binding pathway to GLIC via a membrane-embedded tunnel using an intrasubunit protein lumen as the conduit, an observation that explains the Meyer-Overton hypothesis, or why the lipophilicity of an anesthetic and its potency are generally proportional. Moreover, employing high concentrations of ligand led to the identification of a second transmembrane site (TM2) that inhibits dissociation of anesthetic from the TM1 site and is consistent with the high concentrations of anesthetics required to achieve clinical effects. Finally, asymmetric binding patterns of anesthetic to the channel were found to promote an iris-like conformational change that constricts and dehydrates the ion pore, creating a 13.5 kcal/mol barrier to ion translocation. Together with previous studies, the simulations presented herein demonstrate a novel anesthetic binding site in GLIC that is accessed through a membrane-embedded tunnel and interacts with a previously known site, resulting in conformational changes that produce a non-conductive state of the channel.
Keywords: anesthetic; computational biology; ion channel; membrane protein; molecular dynamics.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
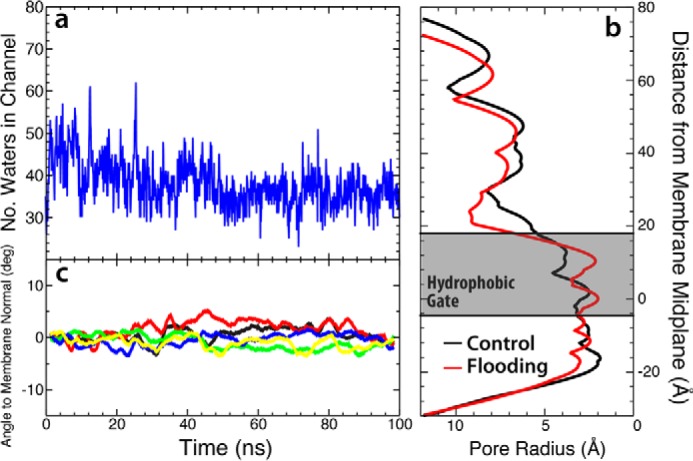
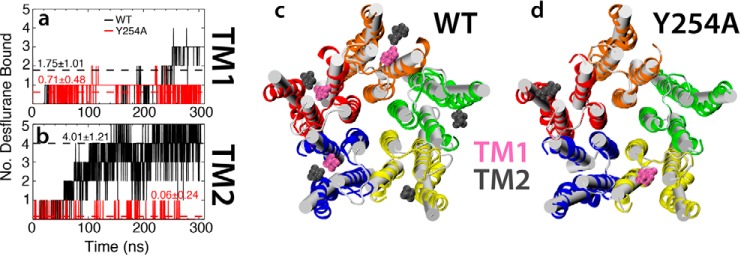
References
-
- Meyer H. H. (1899) The Theory of Alcohol Anesthesia – First Communication. What property of anesthetics causes its narcotic effect? Arch. Exp. Pathol. Pharmakol. 42, 109–118
-
- Seeman P. (1972) The membrane actions of anesthetics and tranquilizers. Pharmacol. Rev. 24, 583–655 - PubMed
-
- Thompson A. J., Lester H. A., and Lummis S. C. (2010) The structural basis of function in Cys-loop receptors. Q. Rev. Biophys. 43, 449–499 - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
