

Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases
- PMID: 28422132
- PMCID: PMC5533198
- DOI: 10.1038/ejhg.2017.49
Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases
Abstract
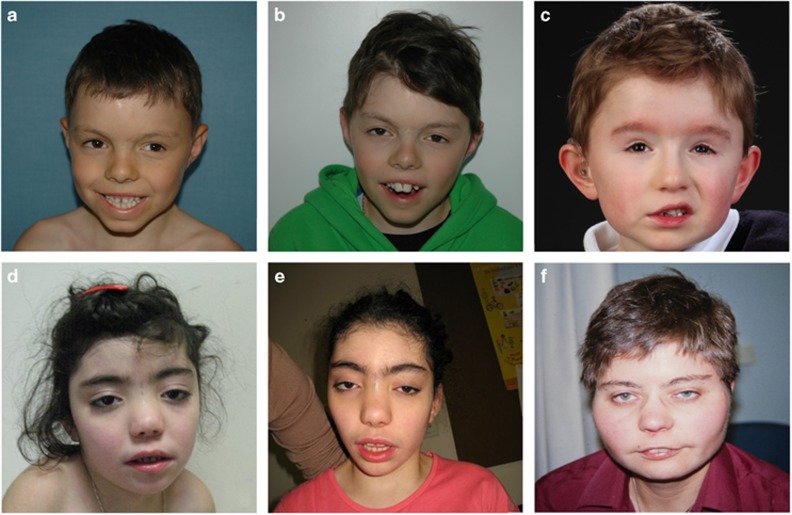
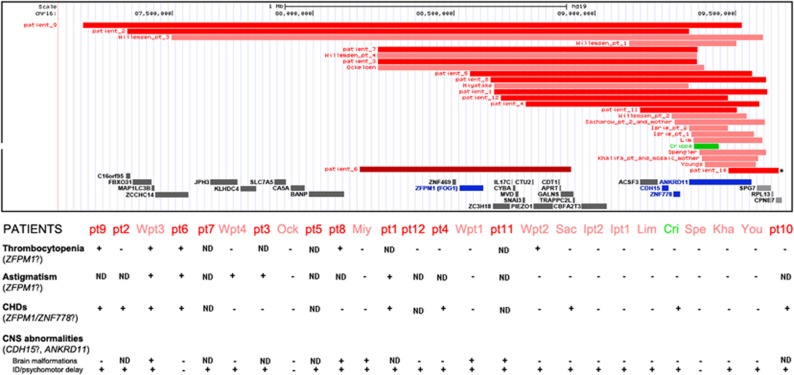
16q24 deletion involving the ANKRD11 gene, ranging from 137 kb to 2 Mb, have been associated with a microdeletion syndrome characterized by variable cognitive impairment, autism spectrum disorder, facial dysmorphisms with dental anomalies, brain abnormalities essentially affecting the corpus callosum and short stature. On the other hand, patients carrying either deletions encompassing solely ANKRD11 or its loss-of-function variants were reported in association with the KBG syndrome, characterized by a very similar phenotype, including mild-to-moderate intellectual disability, short stature and macrodontia of upper incisors, with inter and intrafamilial variability. To assess whether the haploinsufficiency of ANKRD11-flanking genes, such as ZFPM1, CDH15 and ZNF778, contributed to either the severity of the neurological impairment or was associated with other clinical features, we collected 12 new cases with a 16q24.2q24.3 deletion (de novo in 11 cases), ranging from 343 kb to 2.3 Mb. In 11 of them, the deletion involved the ANKRD11 gene, whereas in 1 case only flanking genes upstream to it were deleted. By comparing the clinical and genetic features of our patients with those previously reported, we show that the severity of the neurological phenotype and the frequency of congenital heart defects characterize the deletions that, besides ANKRD11, contain ZFPM1, CDH15 and ZNF778 as well. Moreover, the presence of thrombocytopenia and astigmatism should be taken into account to distinguish between 16q24 microdeletion syndrome and KBG syndrome. The single patient not deleted for ANKRD11, whose phenotype is characterized by milder psychomotor delay, cardiac congenital malformation, thrombocytopenia and astigmatism, confirms all this data.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome.Am J Med Genet A. 2013 Apr;161A(4):835-40. doi: 10.1002/ajmg.a.35739. Epub 2013 Mar 12. Am J Med Genet A. 2013. PMID: 23494856
-
Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome.Am J Med Genet A. 2012 Mar;158A(3):547-52. doi: 10.1002/ajmg.a.34436. Epub 2012 Feb 3. Am J Med Genet A. 2012. PMID: 22307766
-
Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11.Am J Med Genet A. 2016 Nov;170(11):2847-2859. doi: 10.1002/ajmg.a.37878. Epub 2016 Sep 8. Am J Med Genet A. 2016. PMID: 27605097
-
Audiological findings in a de novo mutation of ANKRD11 gene in KBG syndrome: Report of a case and review of the literature.Int J Pediatr Otorhinolaryngol. 2017 Dec;103:109-112. doi: 10.1016/j.ijporl.2017.10.017. Epub 2017 Oct 12. Int J Pediatr Otorhinolaryngol. 2017. PMID: 29224748 Review.
-
Deletion of first noncoding exon in ANKRD11 leads to KBG syndrome.Am J Med Genet A. 2023 Apr;191(4):1044-1049. doi: 10.1002/ajmg.a.63119. Epub 2023 Jan 11. Am J Med Genet A. 2023. PMID: 36628575 Review.
Cited by
-
A case of prenatal diagnosis of 16q24.3 microdeletion KBG syndrome and review of the literature.Clin Case Rep. 2022 Jun 19;10(6):e5958. doi: 10.1002/ccr3.5958. eCollection 2022 Jun. Clin Case Rep. 2022. PMID: 35765297 Free PMC article.
-
Olfactory bulb anomalies in KBG syndrome mouse model and patients.BMC Med. 2024 Apr 15;22(1):158. doi: 10.1186/s12916-024-03363-6. BMC Med. 2024. PMID: 38616269 Free PMC article.
-
A Gene-Based Algorithm for Identifying Factors That May Affect a Speaker's Voice.Entropy (Basel). 2023 Jun 2;25(6):897. doi: 10.3390/e25060897. Entropy (Basel). 2023. PMID: 37372241 Free PMC article.
-
Deep phenotyping of the neuroimaging and skeletal features in KBG syndrome: a study of 53 patients and review of the literature.J Med Genet. 2023 Nov 27;60(12):1224-1234. doi: 10.1136/jmg-2023-109141. J Med Genet. 2023. PMID: 37586838 Free PMC article. Review.
-
16q24.3 Microdeletions Disrupting Upstream Non-Coding Region of ANKRD11 Cause KBG Syndrome.Genes (Basel). 2025 Jan 24;16(2):136. doi: 10.3390/genes16020136. Genes (Basel). 2025. PMID: 40004465 Free PMC article.
References
-
- Skjei KL, Martin MM, Slavotinek AM: KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am J Med Genet A 2007; 143A: 292–300. - PubMed
-
- Khalifa M, Stein J, Grau L et al: Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am J Med Genet A 2013; 161A: 835–840. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
